Fig. 9.1
Abnormal protein misfolding and aggregation as the common molecular pathogenesis of neurodegenerative diseases. Most of the genetic mutations responsible for the inherited neurodegenerative diseases commonly render the mutant proteins prone to misfold and aggregate, or result in the overproduction of aggregation-prone proteins, leading to their accumulation as inclusion bodies in the brain, eventually resulting in neurodegeneration. These facts strongly indicate that abnormal protein misfolding and aggregation cause neurodegeneration through common pathogenic mechanisms, and hence they are called protein misfolding diseases
One of the striking features shared by these protein aggregates is that they have common β-sheet-rich amyloid fibrillar structures, despite their distinct primary amino acid sequences. Accumulating evidence now suggests that rather than the insoluble mature amyloid fibrils, soluble pre-fibrillar intermediates of these proteins, such as β-sheet-rich misfolded monomers, oligomers, or protofibrils, are more toxic (Haass and Selkoe 2007). Furthermore, various experimental animal models expressing these mutant proteins have been established, using mice, flies, and worms, and have been proven to induce progressive neurodegeneration accompanied with protein aggregates/inclusions in the brain, faithfully recapitulating pathological features of the human diseases. These facts, taken together, strongly indicate that abnormal protein misfolding and aggregation cause neurodegeneration by common pathogenic mechanisms in these neurodegenerative diseases, and hence they are called conformational diseases or protein misfolding diseases (Fig. 9.1) (Carrell and Lomas 1997; Dobson 2003).
Although the detailed mechanisms by which these misfolded protein oligomers/aggregates cause neurodegeneration have remained unclear, the dysfunction of various cellular and neuronal processes has been proposed as being responsible for neurodegeneration, including mitochondrial dysfunction, endoplasmic reticulum stress, oxidative stress, inefficient protein degradation, transcriptional dysregulation, intracellular and axonal transport impairment, synaptic dysfunction, excitotoxicity, Ca2+ dysregulation, membrane damage, and the activation of caspases. Accordingly, various disease-modifying therapeutic approaches targeting each of these affected processes are currently being investigated. Among these therapeutic targets, protein misfolding and aggregation are considered to be the most ideal therapeutic targets since they are the earliest events in the pathogenic cascade, and hence inhibition of misfolding/aggregation is expected to widely suppress a broad range of downstream pathogenic changes. In this chapter, various emerging disease-modifying therapeutic approaches for neurodegenerative diseases, particularly those targeting the misfolding and aggregation of toxic proteins are introduced and reviewed. Problems that need to be solved during the development of molecular-targeted disease-modifying therapies are discussed, as are the future prospects of research in this area.
9.2 Alzheimer’s Disease
Alzheimer’s disease (AD) is the most common cause of degenerative dementia, which is pathologically characterized by senile plaques composed of Aβ and neurofibrillary tangles (NFTs) composed of tau. The prevalence of AD is estimated to be more than 30 million people worldwide (5 million people in the United States), and the prevalence increases with age, being 5% of people over 65-years-old and about 30% of people over 85-years-old (Hebert et al. 2013). The estimated annual health-care cost was approximately $200 billion in the United States in 2012, and hence AD is a serious threat to human beings and countermeasures are urgently anticipated.
Since the discovery of mutations in either the APP, PS1, or PS2 gene in familial AD (FAD), most of which overproduce aggregation-prone Aβ42, the “amyloid hypothesis” considering the deposition of Aβ in the brain as the primary cause of AD pathogenesis, has been widely accepted (Hardy and Selkoe 2002; Hardy and Higgins 1992). Accordingly, a large amount of effort put into developing potential therapies for AD has focused on targeting the Aβ deposition. Other therapeutic approaches have also been investigated, for example those targeting tau, which is a major component of the NFTs that are believed to form as a downstream event of Aβ deposition. Although these therapeutic candidates still need to be validated in clinical trials, various promising approaches to combat this devastating disorder are in development as shown below (Table 9.1) (Ghezzi et al. 2013; Nygaard 2013).
Table 9.1
Potential disease-modifying therapies in clinical development for Alzheimer’s disease
Drug | Stage | Outcome | Findings |
|---|---|---|---|
γ-Secretase inhibitors | |||
Semagacestat | Phase III | Terminated | Worsening, skin cancers and infections |
Avagacestat | Phase II | Terminated | Worsening |
β-Secretase inhibitors | |||
LY2886721 | Phase II/III | Terminated | Live toxicity |
MK-8931 | Phase II/III | Ongoing | |
Active immunization | |||
AN1792 | Phase II | Terminated | Meningoencephalitis |
CAD106 | Phase II | Ongoing | |
Passive immunization | |||
Bapineuzumab | Phase III | Completed | Low efficacy |
Solanezumab | Phase III | Completed | Low efficacy |
Gantenerumab | Phase III | Ongoing | |
Crenezumab | Phase II | Ongoing | |
Aβ aggregation inhibitors | |||
Tramiprosate | Phase III | Completed | Low efficacy |
PBT2 | Phase II | Completed | Low efficacy |
Scyllo-inositol | Phase II | Completed | Low efficacy |
Tau aggregation inhibitors | |||
Methylene blue | Phase II | Completed | |
9.2.1 Reduction of Amyloid-β Production: γ- and β-Secretase Inhibitors
Aβ is produced by the sequential cleavage of its precursor APP by two enzymes, namely β-secretase and γ-secretase (Fig. 9.2). β-secretase first cleaves APP at the extracellular cleavage site, and then γ-secretase cleaves the resultant C-terminal APP fragment within the transmembrane domain to generate several Aβ species of 36-43 amino acids. Among the Aβ peptides, the 40-amino-acid peptide (Aβ40) is the most abundant species, whereas the 42-amino-acid peptide (Aβ42) is less abundant but has a higher aggregation propensity. APP is also known to be cleaved by a third enzyme, α-secretase, within the Aβ sequence, resulting in no production of Aβ. Therefore, β-secretase and γ-secretase are considered as rational therapeutic targets to reduce Aβ production (Bignante et al. 2013).
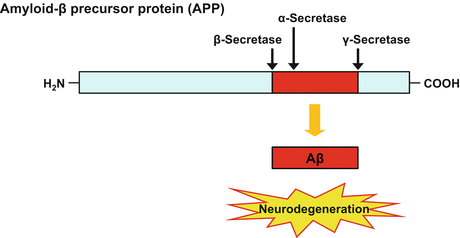
Fig. 9.2
Production of the amyloid-β peptide by cleavage from Aβ precursor protein. Aβ is produced by the sequential cleavage of APP by β-secretase, followed by γ-secretase. APP is also known to be cleaved within the Aβ sequence by α-secretase, resulting in no production of Aβ
9.2.1.1 γ-Secretase Inhibitors
γ-Secretase is a protease complex consisting of nicastrin, Aph-1, and Pen-2 in addition to presenilin, whose mutations cause FAD (Francis et al. 2002). Since γ-secretase is involved in the intramembranous cleavage of not only APP, but also other essential substrate proteins, such as Notch, ErbB4, and N-cadherin, simple inhibition of its enzymatic activity would possibly cause detrimental effects. Indeed, PS1 knockout mice were shown to result in embryonic lethality, with a deformed skeleton and impaired neurogenesis, owing to a defect of the Notch signaling pathway (Shen et al. 1997).
The first γ-secretase inhibitor that was developed and extensively evaluated in clinical studies was Semagacestat (LY450139), which was shown to decrease the Aβ production in the CNS and Aβ concentrations in plasma. Large Phase III trials assessing the clinical efficacy of Semagacestat in AD patients were terminated before their completion due to the worsening of cognitive function as well as other adverse effects, such as skin cancers and infections (Doody et al. 2013). These undesired events were thought to result from the inhibition of Notch signaling. Therefore, γ-secretase inhibitors that specifically inhibit the APP cleavage and spare the Notch cleavage, or the so-called γ-secretase modulators that only shift the γ-secretase cleavage position to produce the less aggregation-prone Aβ40 without affecting the Notch cleavage, have been extensively investigated. Although Avagacestat (BMS-708163), a γ-secretase inhibitor with a minimal effect on Notch signaling, showed a good safety and tolerability profile, and reduced the cerebrospinal fluid (CSF) concentrations of Aβ in a Phase I trial, a Phase II trial in AD patients again showed a trend toward clinical worsening and was thus terminated (Coric et al. 2012). The investigation of other Notch-sparing γ-secretase inhibitors, such as Begacestat (GSI-953) and CHF-5074, are still ongoing (http://www.clinicaltrial.gov).
9.2.1.2 β-Secretase Inhibitors
β-Secretase, also referred to as β-site APP cleaving enzyme 1 (BACE1), is an aspartyl protease, which initiates Aβ production by cleaving the extracellular domain of APP, in competition with α-secretase, before the intramembranous cleavage of the C-terminal fragments (Vassar et al. 1999). Although β-secretase also has other substrates, such as neuregulin 1, BACE1 knockout mice were initially reported to be viable and to show normal development and behavior with no overt phenotype, in contrast to PS1 knockout mice (Luo et al. 2001). Most importantly, BACE1 deficiency in APP transgenic mice has been shown to result in a dramatic reduction of cerebral Aβ levels and a rescue from Aβ-dependent memory deficits (Ohno et al. 2004), indicating BACE1 inhibition as a promising therapeutic strategy for AD without serious adverse effects.
After the successive failures of γ-secretase inhibitors in clinical trials, β-secretase inhibitors became the next focus of attention, as they were expected to have fewer off-target effects. LY2886721 is an orally available BACE1 inhibitor, which was shown to reduce cerebral Aβ concentrations in preclinical animal models. However, a Phase II trial of LY2886721 in patients with mild cognitive impairment or mild AD was recently terminated due to abnormalities in liver biochemical tests, although its administration resulted in dose-dependent decreases in both plasma and CSF Aβ40 concentrations. MK-8931 is a potent β-secretase inhibitor, which was shown to reduce Aβ levels in the CSF and brains of rodents and primates. Following the Phase I trial demonstrating the general safety of MK-8931 and its significant effect on decreasing CSF Aβ levels, two large Phase II/III randomized control trials (RCTs) are currently ongoing for mild-to-moderate AD patients.
9.2.2 Removal of Amyloid-β: Active and Passive Immunization
Various immunological therapeutic approaches against Aβ, including both active and passive immunization, have been actively investigated (Lannfelt et al. 2014). Antibodies against Aβ are thought to enter the brain to some extent, and to remove Aβ from preexisting amyloid plaques via microglial activation and subsequent phagocytosis (Bard et al. 2000). Aβ immunotherapy is also expected to act in the peripheral circulation to reduce Aβ levels, leading to the excretion of monomeric Aβ from the brain according to the equilibrium of Aβ between the peripheral blood and CNS, which is called the “peripheral sink hypothesis” (DeMattos et al. 2001). Aβ antibodies are also expected to directly interfere with the aggregation and further accumulation of Aβ in the CNS. The active immunization approach offers many advantages over passive immunization, in particular, a sustained antibody response by a limited number of vaccinations, and its lower cost. Its potential disadvantage is the variability in the antibody response among patients, including the occurrence of unexpected responses. On the other hand, the potential advantages of passive immunization are the reproducible and reliable delivery of therapeutic antibodies to the patient, and their rapid clearance. This also results in the disadvantage of the requirement for repeated antibody infusions over extended periods.
9.2.2.1 Active Immunization
The potential of Aβ immunotherapy for AD was first demonstrated by Schenk et al. in 1999. They vaccinated APP transgenic mice with Aβ itself, and found a significant reduction of cerebral Aβ plaques, and an amelioration of cognitive impairment (Schenk et al. 1999). However, the first Phase II clinical trial of active immunization using the AN1792 vaccine in AD patients was terminated because 18 of the 298 patients developed meningoencephalitis after 2–3 injections (Gilman et al. 2005). Long-term follow-up through clinical and postmortem neuropathological examinations performed on some of the patients, demonstrated a reduction of brain Aβ plaques in the immunized patients, but no significant differences in cognitive function between antibody responders and the placebo group (Holmes et al. 2008). These results clearly indicate the promising potential of Aβ immunotherapy to remove Aβ plaques, but also suggest that plaque removal may not prevent the progressive cognitive decline in AD.
Recently, a new vaccine called CAD106 was developed, which presents the N-terminal Aβ1-6 peptide epitope to drive a B-cell response and to avoid the T-cell activation that is responsible for meningoencephalitis. CAD106 was shown to successfully reduce Aβ deposition in APP transgenic mice. A Phase I clinical trial of CAD106 found it to be safe and well tolerated, with indications of an antibody response to Aβ, a reduction in serum Aβ levels, and no signs of meningoencephalitis (Winblad et al. 2012).
9.2.2.2 Passive Immunization
Another solution to avoid undesired T cell-induced inflammation that can cause side effects, is to use passive immunization with humanized antibodies. The humanized monoclonal anti-Aβ antibody bapineuzumab, which binds both soluble and insoluble fibrillar Aβ, was shown to reduce the amyloid burden in APP transgenic mice (Bard et al. 2000). This was the first antibody to be tested in clinical trials, and was shown to be generally safe and well tolerated in Phase I/II studies (Wyeth, Madison, NJ, USA, and Élan Corporation, Dublin, Ireland). Recently, two large multicenter, randomized, double-blind, and placebo-controlled Phase III trials were conducted to assess the clinical efficacy of bapineuzumab in mild-to-moderate AD patients. In these trials, the intravenous administration of bapineuzumab resulted in reduced Aβ loads in the brain, detected by positron emission tomography using Pittsburgh compound B (PIB-PET) and reduced phosphorylated tau levels in the CSF of AD patients carrying the ApoE4 allele. However, this treatment failed to improve clinical outcomes, such as cognitive function, and also failed to meet the primary clinical endpoints in these patients (Salloway et al. 2014).
Solanezumab (LY2062430; Eli Lilly) is a newer humanized monoclonal antibody, which binds to an epitope in the central region of Aβ and preferentially targets soluble Aβ oligomers. Solanezumab treatment was shown to increase plasma Aβ concentrations, and to reverse memory deficits in APP transgenic mice (Dodart et al. 2002). In a Phase II trial, the safety and tolerability of solanezumab were shown, together with a dose-dependent increase in plasma and CSF Aβ levels. Two subsequent large Phase III clinical trials of solanezumab in patients with mild-to-moderate AD, however, failed to slow the cognitive decline, although CSF Aβ levels were increased after the treatment (Doody et al. 2014). Pooled data from both trials in mild AD patients revealed a significant slowing of cognitive decline after 78 weeks of treatment, indicating that treatment should be started earlier in the disease process for the maximum beneficial outcome. Eli Lilly and Co. has started a third Phase III clinical trial of solanezumab in patients with mild AD, in which they are also analyzing amyloid depositions by PET.
Gantenerumab (R1450; Hoffman-LaRoche) is a novel human anti-Aβ antibody that has been optimized for high affinity and preferential binding to a conformational epitope expressed on Aβ fibrils using phage display technology (Bohrmann et al. 2012). Gantenerumab treatment significantly reduced the brain Aβ plaques, and prevented new plaque formation without affecting plasma Aβ levels in APP/PS2 transgenic mice. A large Phase III clinical trial of gantenerumab is currently underway.
Crenezumab (MABT5102A; Genentech Inc.) is a novel human anti-Aβ antibody with an IgG4 backbone rather than an IgG1 backbone, which is expected to have a milder effect on microglia and should thus avoid side effects, such as microhemorrhages and vasogenic edema. Genentech and the Banner Alzheimer’s Institute, together with the National Institutes of Health (NIH) have started a prevention trial of Crenezumab in the large Colombian AD family with a PS1 mutation.
9.2.3 Amyloid-β Aggregation Inhibitors
According to the “amyloid hypothesis”, the aggregation and subsequent deposition of Aβ peptides as amyloid plaques in the brain are thought to trigger the pathogenic cascade in AD (Hardy and Selkoe 2002; Hardy and Higgins 1992). Conformational changes and aggregation of Aβ are facilitated by its interaction with various biomolecules such as other proteins, lipids, and metals. Therefore, various therapeutic approaches to prevent Aβ aggregation, either by inhibiting its self-assembly, or its interaction with other biomolecules have been investigated as described below (Dasilva et al. 2010).
Soto and colleagues first developed the idea of using Aβ aggregation inhibitors as potential therapies for AD. The core amyloidogenic sequences of Aβ required for its self-assembly are reported to be the hydrophobic residues 16-20 (KLVFF) and residues 25-35. They designed Aβ-based peptide inhibitors, the so-called “β-sheet breaker peptides”, by introducing a single proline substitution in the former core amyloidogenic region to abolish the β-sheet propensity of Aβ (Soto et al. 1996). Τhese β-sheet breaker peptides were indeed shown to inhibit Aβ fibril formation and to disassemble preformed Aβ fibrils in vitro, and furthermore, were shown to reduce amyloid deposition, neuronal loss, and accompanying brain inflammation in Aβ-injected amyloidosis rats, and APP transgenic mice (Soto et al. 1998; Permanne et al. 2002). These studies clearly indicated that Aβ aggregation is one of the causes of neurodegeneration in the pathogenesis of AD, and that the inhibition of Aβ aggregation leads to the suppression of downstream pathogenic events.
Tramiprosate (3-amino-1-propanesulfonic acid/Alzhemed; Neurochem Inc.) is a glycosaminoglycan (GAG) mimetic that interferes with the binding of GAGs with the Aβ peptide, thereby preventing Aβ fibril formation. Tramiprosate treatment was reported to reduce the amyloid plaque load, as well as soluble and insoluble Aβ levels in the brain of APP transgenic mice (Gervais et al. 2007). Although tramiprosate was shown in a Phase II study to be safe and well tolerated, to cross the blood-brain barrier (BBB), and to dose-dependently reduce CSF Aβ levels, a large Phase III clinical trial in mild-to-moderate AD patients failed to meet the predefined primary endpoints. However, post-hoc analyses revealed its positive and significant effects on secondary endpoints, including the attenuation of hippocampal volume loss, as well as a trend toward the slowing of cognitive decline (Aisen et al. 2011), suggesting the potential of tramiprosate as a disease-modifying drug for AD.
PBT2, an 8-hydroxy quinolone derivative, works as a chelator of copper, zinc and iron, and thereby disrupts the aberrant interactions of these metals with Aβ. Treatment of APP/PS1 transgenic mice with PBT2 resulted in a decrease in the brain Aβ levels and cognitive improvements (Adlard et al. 2008). In a Phase IIa clinical trial in early AD patients, the cognitive efficacy of PBT2 was limited to only two measures of executive function, although a dose-dependent and significant reduction in CSF Aβ42 levels was observed (Lannfelt et al. 2008).
Scyllo-inositol (cyclohexanehexol/AZD-103), a derivative of the natural glycolipid phosphatidylinositols, has been shown to interfere with Aβ aggregation by competing with phosphatidylinositol lipids for binding to Aβ, which is known to facilitate Aβ fibril formation. McLaurin and colleagues showed that oral administration of scyllo-inositol reduces brain Aβ levels and amyloid plaques, and improves cognitive deficits in APP transgenic mice (McLaurin et al. 2006). These therapeutic effects were still observed even when mice were treated after the onset of AD-like symptoms. However, a Phase II clinical trial of scyllo-inositol resulted in no evidence of clinical benefit, and only a decrease in the CSF Aβ levels was observed (Salloway et al. 2011). Therefore, the clinical efficacy of scyllo-inositol awaits validation in a larger Phase III study.
Epigallocatechin-3-gallate (EGCG) is a major polyphenolic component of green tea. Although much attention was paid to its anti-oxidant activity against Aβ-induced toxicity in earlier studies, EGCG was subsequently shown to bind directly to Aβ, thereby inhibiting its fibrillization, and instead, redirecting Aβ to assemble into non-toxic large spherical aggregates in vitro (Ehrnhoefer et al. 2008). EGCG also targets other aggregation-prone proteins, such as mutant huntingtin with a polyglutamine expansion, tau, α-Syn, prion, transthyretin (TTR), and islet amyloid polypeptide (IAPP) to inhibit their amyloid fibrillization. Administration of EGCG to APP transgenic mice was reported to reduce Aβ levels and amyloid plaque load, as well as cognitive impairment, indicating its therapeutic potential in vivo (Rezai-Zadeh et al. 2005).
Curcumin is a major constituent of the dietary spice turmeric, which has been reported to exert anti-oxidant and anti-inflammatory properties (Monroy et al. 2013). Curcumin was found to inhibit Aβ fibril formation and also to destabilize preformed Aβ fibrils in vitro (Ono et al. 2004). It was shown to bind directly to the fibrillar conformation of Aβ, and to also inhibit the aggregation of other amyloidogenic proteins including Aβ, α-Syn, prion, TTR and IAPP. In vivo studies showed that peripherally-injected curcumin crosses the BBB and labels amyloid plaques in APP transgenic mice, and its administration reduced amyloid plaque burden (Yang et al. 2005). In a Phase II clinical trial in mild-to-moderate AD patients, oral administration of curcumin was shown to be generally safe and well tolerated, but it failed to show any clinical or biomarker efficacy (Ringman et al. 2012), and the plasma curcumin levels were found to be very low. The major obstacle to the clinical application of curcumin is its poor bioavailability, mainly due to its insolubility, poor absorption, and rapid metabolism (Anand et al. 2007). Therefore, derivatives of curcumin with improved bioavailability and delivery, are highly anticipated due their role as potential therapeutic agents against these incurable diseases in the near future.
9.2.4 Degradation of Amyloid-β
Since the aggregation and subsequent deposition of Aβ in the brain are regarded as the primary culprits in the pathogenesis of AD (Hardy and Selkoe 2002), the degradation of Aβ, and not only its production, is thought to be one of the more attractive therapeutic targets for AD. In particular, a recent report showing that the clearance of Aβ, rather than its production, is altered in AD patients, has highlighted the importance of Aβ degradation (Mawuenyega et al. 2010). Although there are many enzymes capable of cleaving Aβ in vitro, only a few have been confirmed to be involved in degrading Aβ in vivo.
Neprilysin, a zinc-dependent metalloprotease, is the most extensively studied Aβ-degrading enzyme in the brain (Iwata et al. 2000). Genetic disruption of the neprilysin gene in mice confirmed its importance in the degradation of endogenous Aβ, as well as exogenously administered Aβ (Iwata et al. 2001). On the other hand, expression of neprilysin in APP transgenic mice, either by crossing them with neprilysin transgenic mice or by viral vector-mediated gene transfer, has been shown to significantly reduce brain Aβ levels and amyloid plaques, and to improve their life span (Leissring et al. 2003; Marr et al. 2003), indicating its therapeutic potential. It is noteworthy that the effect of environmental enrichment on reducing Aβ load in APP mice is closely correlated with the upregulation of neprilysin levels (Lazarov et al. 2005).
Insulin-degrading enzyme (IDE), a thiol metalloendopeptidase that degrades small peptides such as insulin and glucagon, is another protease that plays an important role in degrading Aβ in the brain (Qiu et al. 1998). Indeed, IDE knockout mice exhibited increased levels of endogenous Aβ in the brain, and developed hyperinsulinemia and glucose intolerance, which have also been proven to be risk factors for AD (Farris et al. 2003). The overexpression of IDE as well as neprilysin, has been shown to reduce the Aβ load and associated brain changes in APP transgenic mice (Leissring et al. 2003). Therefore, the discovery of these Aβ-degrading enzymes has revealed novel therapeutic opportunities for AD through the upregulation of their activities.
9.2.5 Tau Aggregation Inhibitors
From the failures of most Aβ-targeted disease-modifying therapies in demonstrating clinical efficacy for AD patients as described above, two major concerns have emerged. First, the time point of treatment initiation might be too late in the course of the disease. In most preclinical studies, therapeutic interventions were started before disease onset in genetically-engineered animals destined to develop the disease. On the other hand, AD-related pathological changes are considered to begin many years before the clinical onset of symptoms (Price and Morris 1999; Bateman et al. 2012), which might prevent the translation of preclinical results obtained from animal models, to clinical efficacy in human AD patients. Another concern is the validity of the therapeutic target. Notably, although Aβ immunotherapies have succeeded in the prevention and removal of amyloid deposition in AD patients, they did not lead to an improvement of cognitive decline. While both amyloid plaques and NFTs are the characteristic pathological features of AD, NFT pathology is known to be more closely correlated with the severity of clinical symptoms of AD than amyloid plaques (Arriagada et al. 1992). Furthermore, the reduction of endogenous tau levels was reported to ameliorate cognitive deficits without affecting the Aβ pathology in APP transgenic mice (Roberson et al. 2007). These facts challenge the current amyloid cascade hypothesis for AD pathogenesis. In addition, the discovery of tau mutations in frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP-17), another form of degenerative dementia, highlighted the significant role of tau in the pathogenesis, which is solely sufficient to cause neurodegeneration in the absence of amyloid pathology (Hutton et al. 1998; Spillantini et al. 1998). Therefore, an alternative therapeutic target, namely tau, the key component of NFTs, is currently being considered and intensively investigated (Giacobini and Gold 2013).
Tau is a member of the microtubule-associated protein family that is involved in the assembly and stabilization of microtubules. It is predominantly expressed in neurons, particularly in their axons, to maintain neuronal morphology and axonal transport. Six tau isoforms are produced via alternative mRNA splicing, which mainly differ in their number of three or four microtubule binding repeats, and all are expressed in the adult human brain. In the AD brain, tau is abnormally hyperphosphorylated and aggregated into NFTs. Hyperphosphorylation of tau is known to reduce the binding ability of tau to microtubules, similar to its FTDP-linked mutations (Hong et al. 1998). However, whereas the hyperphosphorylation of tau is reported to promote its self-assembly into NFTs (Alonso et al. 2001), tau itself can assemble into β-sheet-rich filaments through its microtubule binding repeat. Among the various aggregated species, tau oligomers are believed to be toxic, leading to synaptic dysfunction (Lasagna-Reeves et al. 2011). Thus, therapeutic approaches to inhibit tau aggregation are currently being investigated.
The first tau aggregation inhibitor reported was the phenothiazine methylene blue (MB), which blocks tau-tau interactions through the microtubule-binding repeat domain (Wischik et al. 1996). Administration of MB was shown to reduce the soluble tau levels, but not pre-existing NFTs, resulting in an improvement of cognitive deficits in tau transgenic mice (O’Leary et al. 2010). A Phase II clinical trial of MB in mild-to-moderate AD patients was conducted and reported to have beneficial effects (Schirmer et al. 2011). Detailed reports on this Phase II study are anticipated, and Phase III trials with a new form of this compound are now underway (Wischik et al. 2014).
9.2.6 Tau Immunotherapy
Recent studies have indicated that tau aggregates can be secreted from neurons, and extracellular tau aggregates contribute to the neuron-to-neuron propagation of tau pathology via a prion-like transmission mechanism (Clavaguera et al. 2009; Frost et al. 2009). Therefore, therapeutic approaches to remove extracellular tau aggregates by immunotherapy are under investigation.
The first active immunization study using recombinant human tau with no phosphorylation resulted in the induction of tauopathy-like pathologies and encephalomyelitis with neurological deficits in mice, indicating the potential risk of using unphosphorylated tau to elicit an autoimmune response (Rosenmann et al. 2006). However, subsequent active immunization studies using phosphorylated tau have succeeded in reducing tau pathology and improving cognitive deficits in mutant tau transgenic mice (Asuni et al. 2007). Passive immunization studies using either phosphorylated tau antibodies or a conformation-dependent tau antibody, have also demonstrated significant improvements in motor deficits and tau pathology in mutant tau transgenic mice (Boutajangout et al. 2011; Chai et al. 2011). Taken together, these results indicate the potential of tau immunotherapy as a therapeutic approach for AD and related tauopathies.
9.2.7 Tau Phosphorylation Inhibitors
The hyperphosphorylation of tau is believed to be one of the most critical steps in tau-mediated neurodegeneration, although it is still controversial as to whether phosphorylation of tau triggers its aggregation, or is just a consequence of its deposition. Therefore, kinases and phosphatases involved in tau phosphorylation and dephosphorylation, respectively, are regarded as therapeutic targets for AD and other tauopathies. Although several kinases, such as glycogen synthase kinase 3 β (GSK-3β), cyclin dependent kinase 5 (CDK5), casein kinase 1 (CK1), and mitogen activated protein kinases (MAPKs) have been reported to phosphorylate tau in vitro, GSK-3β is believed to be the most promising candidate tau kinase because it is activated by Aβ and is essential for Aβ-induced neurotoxicity, thus linking Aβ to tau phosphorylation.
Administration of the GSK-3 inhibitor lithium chloride has been shown to reduce the levels of tau phosphorylation and insoluble aggregation, as well as the degree of axonal degeneration in mutant tau transgenic mice (Noble et al. 2005). Accordingly, small short-term clinical trials of lithium chloride have been conducted in mild-to-moderate AD patients, resulting in no beneficial effect on cognitive impairment, but a significant decrease in CSF phosphorylated tau levels (Forlenza et al. 2011; Hampel et al. 2009). Tideglusib (NP-12), a novel specific GSK-3 inhibitor, was tested in double-transgenic mice co-expressing human mutant APP and tau, resulting in decreased tau phosphorylation and Aβ deposition, as well as the prevention of memory deficits (Sereno et al. 2009). Currently, phase II clinical trials of tideglusib in patients with AD and progressive supranuclear palsy (PSP), one of the tauopathies, are ongoing.
Another approach to inhibit tau hyperphosphorylation is to activate the protein phosphatases that dephosphorylate tau. Protein phosphatase 2A (PP2A) was reported to be the main phosphatase involved in tau phosphorylation in the brain (Goedert et al. 1992; Gong et al. 2000). Sodium selenate was shown to increase PP2A activity, to prevent tau hyperphosphorylation and neurodegeneration, and to improve motor performance in mouse models of tauopathy (van Eersel et al. 2010).
9.3 Parkinson’s Disease
Parkinson’s disease is the second most common neurodegenerative disorder after AD, and is the most common movement disorder that causes progressive motor symptoms such as rest tremor, rigidity, bradykinesia, gait disturbance and postural instability (de Lau et al. Lancet Neurol 2006). It has recently been recognized that patients also exhibit various non-motor symptoms including olfactory deficit, depression, pain, sleep disturbance and autonomic dysfunction (Chaudhuri et al. Lancet Neurol 2009). The pathological hallmark of PD is the selective loss of dopaminergic neurons in the substantia nigra pars compacta, accompanied by Lewy bodies and Lewy neurites, both of which are distinctive intraneuronal inclusions (Lees et al. Lancet 2009). The main component of these inclusions is the misfolded and aggregated form of α-Syn, which is at least partially post-translationally modified by phosphorylation. Treatments currently available for PD are limited to symptomatic therapies such as dopamine replacement, which only partially improves symptoms, and no effective disease-modifying therapy has been developed to date.
Since the discovery of both missense and multiplication mutations in the α-Syn gene as causes of familial PD and dementia with Lewy bodies (DLB), and the identification of single-nucleotide polymorphisms (SNPs) in the α-Syn gene as a risk factor of sporadic PD, α-Syn has been suggested to play a critical role in the pathogenesis of PD and related synucleinopathies. Indeed, experimental overexpression of α-Syn was shown to mimic several aspects of PD in transgenic animals, such as motor dysfunction, α-Syn aggregation/accumulation, and neurodegeneration. Importantly, familial PD-linked mutations in α-Syn were shown to accelerate its aggregation as well as neurotoxicity. Furthermore, aggregated α-Syn has recently been shown to be secreted from cells, and is believed to be transmitted from neuron to neuron via prion-like mechanisms, thus contributing to the progressive spread of pathology (Jucker and Walker 2013). Therefore, similar to the approaches against AD, various therapeutic approaches targeting protein misfolding and aggregation of α-Syn have been investigated against PD and the other synucleinopathies, although most of them are still at preclinical experimental stages.
In 2001, Masliah and colleagues first demonstrated that β-synuclein (β-Syn) acts as an inhibitor of α-Syn aggregation, and that overexpressed β-Syn ameliorated motor deficits and neurodegeneration in α-Syn transgenic mice (Hashimoto et al. 2001), although β-Syn itself was later found to be linked to DLB, and could possibly induce neurotoxicity. El-Agnaf and colleagues designed a synthetic peptide derived from the hydrophobic non-Aβ component domain of α-Syn, which has been shown to prevent α-Syn aggregation and toxicity (El-Agnaf et al. 2004).
Various small chemical compounds, including baicalein, EGCG, curcumin, rifampicin, etc., have been discovered to show inhibitory activities on α-Syn aggregation in vitro, although most of them still remain to be evaluated for their therapeutic effects in vivo (Ono et al. 2008; Ono and Yamada 2006). Among these, the flavinoid compound baicalein was shown not only to inhibit the fibrillation of α-Syn, but also to disaggregate its preformed fibrils (Zhu et al. 2004). EGCG was reported to modify the fibrillogenesis pathway of α-Syn, as in Aβ (Ehrnhoefer et al. 2008). More recently, EGCG was found to delay the climbing dysfunction and to reduce oxidative stress in a PD Drosophila model expressing α-Syn (Siddique et al. 2014a). Curcumin was also shown to delay the decrease in activity, to reduce oxidative stress, and to extend the life span in α-Syn expressing PD flies (Siddique et al. 2014b), suggesting that multiple mechanisms may be involved in its therapeutic effects (Monroy et al. 2013). The macrocyclic antibiotic rifampicin is an inhibitor of α-Syn aggregation (Li et al. 2004) that is currently being developed for clinical evaluation. Rifampicin treatment was reported to suppress α-Syn aggregation and its associated neurodegeneration in a mouse model of multiple system atrophy (MSA), a synucleinopathy, which expresses α-Syn in oligodendrocytes (Ubhi et al. 2008). However, a clinical trial of rifampicin in MSA patients was recently terminated because of no significant clinical efficacy (Low et al. 2014).
As with immunological therapeutic approaches for AD, various immunotherapies targeting α-Syn have been extensively investigated. The first study was reported by Masliah and colleagues, in which vaccination of α-Syn transgenic mice with recombinant human α-Syn resulted in the production of high affinity antibodies accompanied with a decreased accumulation of aggregated α-Syn and reduced neurodegeneration (Masliah et al. 2005). Passive immunization with exogenously administered human α-Syn antibodies has been shown to prevent the neuron-to-astroglia transmission of α-Syn, and to ameliorate neurodegeneration and behavioral deficits in α-Syn expressing mice (Bae et al. 2012). Among the various immunotherapies targeting α-Syn, active immunization by vaccination with AFFITOPE® PD01 (Affiris, Vienna, Austria) is currently being evaluated in a Phase I clinical study for PD patients. This antigen consists of a peptide carrier conjugate, with aluminium hydroxide as the immunological adjuvant, and targets aggregated α-Syn but not β-Syn. AFFITOPE® PD01 has been shown to reduce the level of α-Syn deposits in the brain, to decrease neuronal loss, and to improve the behavioral deficit in the Morris Water Maze test in two independent PD mouse models (Schneeberger et al. 2012).
Phosphorylation of α-Syn has been implicated to be linked to the pathology of PD, because α-Syn is hyperphosphorylated, particularly at serine 129 (Ser129), in Lewy bodies of PD patients and animal models overexpressing α-Syn (Braithwaite et al. 2012). Although several functional consequences of α-Syn phosphorylation have been reported from in vitro studies, the dephosphorylation of α-Syn at Ser129 in α-Syn transgenic mice by the upregulation of phosphoprotein phosphatase 2A (PP2A), was associated with reduced α-Syn aggregation, improved dendritic arborization and neuronal activity, and amelioration of motor deficits (Lee et al. 2011).
Although our understanding of the pathogenesis of PD has greatly increased in the past 20 years, no agents have yet been proved to alter its disease course to stop or slow its clinical progression. Further research toward the development of disease-modifying therapies for PD is anticipated.
9.4 Amyotrophic Lateral Sclerosis
Amyotrophic lateral sclerosis (ALS) is the most common adult form of motor neuron disease, affecting the upper and lower motor neurons in the brain and spinal cord. Patients develop progressive muscle weakness, typically leading to death, mainly due to respiratory failure within three to five years from disease onset. Similar to AD and PD, a key pathological characteristic of ALS is the presence of cytoplasmic inclusions/aggregates in degenerating motor neurons. The predominant form of aggregates are ubiquitinated and classified as Lewy body-like inclusions, while ubiquitin-negative eosinophilic inclusions called Bunina bodies, are also found.
Although SOD1 was the first gene identified as a causative gene for familial ALS (FALS), mutations in the SOD1 gene account for only 20% of FALS patients, and the inclusions found in the vast majority of sporadic and FALS patients are SOD1-negative. Nevertheless, most SOD1 mutants were found to be prone to misfold and aggregate, which is associated with neurotoxicity in cellular and animal models, and to be accumulated in SOD1-linked FALS patients, suggesting that these diseases can also be categorized as protein misfolding diseases (Shaw and Valentine 2007). Lansbury and colleagues performed in silico screening for small molecules that would stabilize the SOD1 dimer. They successfully identified 15 chemical compounds that inhibit the aggregation of FALS-linked SOD1 mutants in vitro, as potential lead compounds against FALS (Ray et al. 2005).
A significant breakthrough was achieved in 2006, when TDP-43 was identified as a major component of the ubiquitinated inclusions in the majority of patients with sporadic ALS, and tau-negative frontotemporal lobar degeneration (FTLD) (Neumann et al. 2006). Subsequently, mutations in the TDP-43 gene were identified as genetic causes of FALS (Sreedharan et al. 2008). Importantly, TDP-43 has a glutamine/asparagine-rich domain with a strong propensity to aggregate at the C-terminus, where most FALS-linked mutations are located, and indeed, mutations in TDP-43 were shown to accelerate its aggregation (Guo et al. 2011; Johnson et al. 2009). These facts strongly indicate that aggregation of TDP-43 is the initial event in the pathogenic cascade of ALS and FTLD, which are collectively called the TDP-43 proteinopathies.
Research toward counteracting TDP-43 aggregation using similar approaches to those used in AD and PD has been initiated in the past several years, and their therapeutic effects still remain to be evaluated in in vivo models. Methylene blue (MB) and dimebon have been reported to reduce the formation of TDP-43 aggregates in cell culture (Yamashita et al. 2009). However, the administration of MB has failed to show any protective effects in two transgenic mouse models of ALS expressing either mutant SOD1 (G93A) or mutant TDP-43 (G348C) (Audet et al. 2012). Further intensive research toward understanding the molecular basis of ALS will hopefully lead to the establishment of effective therapeutic strategies to overcome this devastating disease in the near future.
9.5 Polyglutamine Diseases
The polyglutamine (polyQ) diseases are a group of inherited neurodegenerative diseases, and are caused in common by an expansion mutation of the glutamine-encoding CAG repeat (>35–40 repeats) in the various disease-causative genes. So far nine diseases, including Huntington’s disease (HD), spinocerebellar ataxia (SCA) type 1, 2, 3, 6, 7 and 17, dentatorubral pallidoluysian atrophy (DRPLA), and spinobulbar muscular atrophy (SBMA), have been found to belong to this group (Table 9.2) (Bauer and Nukina 2009; Nagai and Popiel 2008; Orr and Zoghbi 2007). It is important to note that the disease-causative proteins have neither sequence homology nor any functional similarities, except for the polyQ stretch itself, the size of which tightly correlates with disease severity. Furthermore, these diseases (except for SBMA) are inherited in an autosomal dominant manner. These unique genetic characteristics strongly indicate that the polyQ diseases are caused by gain of toxic function mechanisms triggered by the expanded polyQ stretch itself (Nagai and Popiel 2008). Indeed, expression of the expanded polyQ peptide alone, or a polyQ stretch fused with non-native proteins, has been proven to cause neurodegeneration in experimental animal models.
Table 9.2
The polyglutamine diseases
Diseases | Gene | CAG repeat | Affected regions | |
|---|---|---|---|---|
Normal | Disease | |||
Spinobulbar muscular atrophy (SBMA)/Kennedy’s disease | androgen receptor | 9–36 | 38–65 | Brainstem, spinal cord |
Huntington’s disease (HD) | huntingtin | 6–35 | 36–180 | Caudate nucleus, putamen, cerebral cortex |
Spinocerebellar ataxia type 1 (SCA1) | ataxin-1 | 6–39 | 39–83 | Cerebellar cortex, dentate nucleus, brainstem, cerebral cortex |
Spinocerebellar ataxia type 2 (SCA2) | ataxin-2 | 14–32 | 32–200 | Cerebellar cortex, brainstem, cerebral cortex, peripheral nerves |
Spinocerebellar ataxia type 3 (SCA3)/Machado-Joseph disease | ataxin-3 | 12–41 | 55–84 | Dentate nucleus, basal ganglia, brainstem, spinal cord |
Spinocerebellar ataxia type 6 (SCA6) | α1A calcium channel | 4–19 | 20–33 | Cerebellar cortex |
Spinocerebellar ataxia type 7 (SCA7) | ataxin-7 | 4–35 | 37–306 | Cerebellum, brainstem, retina |
Spinocerebellar ataxia type 17 (SCA17) | TATA-binding protein | 25–44 | 46–63 | Cerebellum, cerebral cortex, basal ganglia |
Dentatorubral-pallidoluysian atrophy (DRPLA) | atrophin-1 | 6–36 | 49–88 | Dentate and red nuclei, cerebellum, cerebral cortex, brainstem |
In the common molecular pathogenesis of the polyQ diseases, expansions of the polyQ stretch were shown to induce misfolding of the host proteins, leading to the formation of insoluble β-sheet-rich aggregates/inclusion bodies in affected neurons, eventually resulting in neurodegeneration. Although it still remains controversial as to whether insoluble inclusion bodies are neurotoxic, protective, or just resultant by-products, soluble β-sheet-rich misfolded monomers and oligomers have been demonstrated to trigger cytotoxicity (Nagai et al. 2007; Schaffar et al. 2004; Takahashi et al. 2008). Therefore, the misfolding and aggregation of expanded polyQ proteins are considered to be ideal therapeutic targets, as they are the most upstream events in the pathogenic cascade, and hence inhibition of misfolding/aggregation is expected to widely suppress various downstream pathogenic events (Herbst and Wanker 2006; Michalik and Van Broeckhoven 2003; Nagai and Popiel 2008).
9.5.1 Intrabodies
Intracellular antibodies, or so-called intrabodies that recognize the expanded polyQ protein have been applied to inhibit its aggregation. The first reported intrabody, C4, was identified from a single-chain Fv phage-display library by Messer and colleagues in 2001, which recognizes the N-terminal region adjacent to the polyQ stretch of huntingtin (Htt). Indeed, they showed that expression of C4 in cell culture potently inhibits the formation of polyQ-expanded mutant Htt inclusions (Lecerf et al. 2001). Furthermore, they successfully demonstrated that the expression of C4 suppresses neurodegeneration in a Drosophila model of HD (Wolfgang et al. 2005), and its viral vector-mediated expression delays Htt aggregation in the brain of HD model mice (Snyder-Keller et al. 2010).
Other intrabodies include VL 12.3, which recognizes the N-terminal region of Htt (Colby et al. 2004), and MW7 and Happ1, both of which recognize the proline-rich region (PRR) of Htt (Khoshnan et al. 2002; Southwell et al. 2008). VL 12.3 was shown to suppress Htt aggregation and cytotoxicity in yeast, although it failed to show any beneficial effects on HD mouse models upon viral vector-mediated expression (Southwell et al. 2009). On the other hand, Happ1 was shown to promote the turnover of mutant Htt, and Happ1 expression successfully suppressed Htt aggregation, and improved motor and cognitive deficits in HD mouse models (Southwell et al. 2008, 2009). Viral vector-mediated expression of another intrabody EM48, which is thought to recognize a C-terminal epitope of human Htt exon1, was also shown to suppress Htt neuropil aggregation and to ameliorate neurological symptoms in a mouse model of HD (Wang et al. 2008).
The application of intrabodies appears to be an attractive approach with regard to their high binding affinity to the disease-causative proteins. However, most of these intrabodies cannot be applied to polyQ diseases other than HD, and may possibly exert unfavorable effects on the normal Htt protein, because intrabodies that specifically recognize the expanded polyQ stretch itself have not been identified so far. The most critical issue is their delivery into the human brain, as they currently require to be expressed via gene delivery due to their large size.
9.5.2 Peptides
Peptides have potential as therapeutic molecules since they are of relatively low molecular weight, and are therefore expected to be delivered to the target organ by their administration (Takeuchi et al. 2014). Indeed, various peptides-based therapeutic approaches have been applied to inhibit protein aggregation, and have been reported to exert therapeutic effects in cell culture and animal models of other neurodegenerative diseases, such as AD and PD (El-Agnaf et al. 2004; Soto et al. 1998).
Based on the hypothesis that peptides with selective binding affinity to the expanded polyQ stretch are expected to interfere with the conformational changes and aggregation of polyQ proteins, we screened combinatorial peptide phage display libraries. We successfully identified polyQ binding peptide 1 (QBP1: SNWKWWPGIFD), which selectively binds to the abnormally expanded polyQ stretch with a dissociation constant of 5.7 μM (Nagai et al. 2000; Okamoto et al. 2009; Popiel et al. 2013), and showed that QBP1 indeed inhibits the toxic β-sheet conformational transition and aggregation of the expanded polyQ protein in vitro (Nagai et al. 2007, 2000). We also found that expression of QBP1 significantly suppresses polyQ oligomer formation and polyQ-induced cytotoxicity in cell culture (Nagai et al. 2000; Takahashi et al. 2008, 2007), and suppresses polyQ-induced neurodegeneration in Drosophila models of the polyQ diseases (Nagai et al. 2003). We further demonstrated that QBP1 conjugated with protein transduction domains (PTD-QBP1) is efficiently delivered into cultured cells, and its oral administration successfully suppresses polyQ-induced premature death in Drosophila, indicating the potential of PTD-QBP1 as a therapeutic molecule (Popiel et al. 2007). We further investigated the therapeutic potential of PTD-QBP1 on a polyQ diseases mouse model; however, repeated intraperitoneal injection of PTD-QBP1 resulted in only a modest improvement of body weight loss, with no improvement in the motor phenotypes, nor suppression of inclusions in the brains, probably due to its low BBB permeability (Popiel et al. 2009). Therefore, one of the future directions is obviously to design low molecular weight chemical analogs of QBP1 with high BBB permeability (Popiel et al. 2013; Tomita et al. 2009).
Related posts:
 Parkinson’s Disease; Neurodegeneration as Systemic Disease
Parkinson’s Disease; Neurodegeneration as Systemic Disease
 Brain–Peripheral Organ Communication
Intermediate Phenotype Approach for Neuropsychiatric Disorders
Brain–Peripheral Organ Communication
Intermediate Phenotype Approach for Neuropsychiatric Disorders
 Clinical Systems Neuroscience
Clinical Systems Neuroscience
 Home- and Community-based Medical Care for Neurodegenerative Diseases: ALS as an Illustration
Home- and Community-based Medical Care for Neurodegenerative Diseases: ALS as an Illustration
 Significance of Mechanism-Oriented Research Toward Neuronal Protection Therapy Against Neurodegenerative Disorders ~ ZNRF1 E3 Ubiquitin Ligase as a Critical Mediator for Wallerian Degeneration and Neuronal Apoptosis
Significance of Mechanism-Oriented Research Toward Neuronal Protection Therapy Against Neurodegenerative Disorders ~ ZNRF1 E3 Ubiquitin Ligase as a Critical Mediator for Wallerian Degeneration and Neuronal Apoptosis
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


