, Damien Gervasoni1 and Catherine Vogt2
(1)
Lyon Neuroscience Research Centre CNRS UMR 5292–INSERM U 1028, Université Claude Bernard Lyon 1, Lyon, France
(2)
Université Claude Bernard Lyon 1, Lyon, France
During the first half of the twentieth century, it seemed impossible to explain the rapidity of interneuronal signal transmission other than by the electric nature of the phenomenon. Experiments carried out in the 1920s by the German physiologist Otto Loewi showed that neurons could transmit electric signals to each other through chemical mediators. Since then, the concept of chemical support of communication between nervous cells has been universally accepted by neurobiologists. This information transfer is afforded by neurotransmitters, chemical substances that fulfill several strict criteria. They are synthesized within the neuron and stored within vesicles in the nerve terminal; they are released by exocytose in a calcium-dependent manner in response to the activation of the nerve terminal; the amount released must be necessary and sufficient to exert an action on the postsynaptic neuron or effector cell; after its release, the neurotransmitter binds to postsynaptic receptors and the remainder is eliminated by mechanisms of inactivation in the synaptic cleft; when the substance is administered exogenously at the appropriate concentrations, it must mimic the action of the endogenous released neurotransmitter. A large number of substances satisfy these criteria, but only a certain number of them will be addressed in this chapter: the amino acids glutamate, gamma-aminobutyric acid (GABA), and glycine and the monoamines noradrenaline, dopamine and serotonin, and acetylcholine (Bjorklund et al. 2005; Cooper et al. 2003; Epelbaum 1995; Meunier and Shvaloff 1995).
3.1 The Amino Acids
The majority of synapses in the brain utilize amino acids as neurotransmitters. The brain contains a large number of amino acids or their derivatives that induce excitation or inhibition when applied to neurons. The three principal amino acid transmitters are glutamate, gamma-aminobutyric acid (GABA), and glycine.
3.1.1 Glutamate
Glutamate (Fig. 3.1) is the principal excitatory neurotransmitter in the CNS. It is involved in phenomena of synaptic plasticity, such as long-term potentiation and trophic roles in development, but also in neuronal excitotoxicity related to excessive calcium influx.
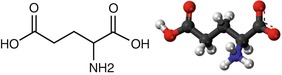
Fig. 3.1
Chemical formula and structure of glutamate
3.1.1.1 Brain Localization
Neurons using glutamate as their neurotransmitter are among the most difficult to identify. In fact, in the brain, glutamate is an amino acid precursor in the synthesis of peptides and proteins, certain of which are neurotransmitters themselves, such as GABA. The identification of glutamatergic neurons is thus problematic in terms of distinguishing between their metabolic and neurotransmitter pools of glutamate. Glutamate is the principal transmitter in most of the efferent and intrinsic projections of the cerebral cortex (Fig. 3.2). It is also found in the thalamocortical projections, in the cerebellar cortex, in the hippocampal formation, etc.
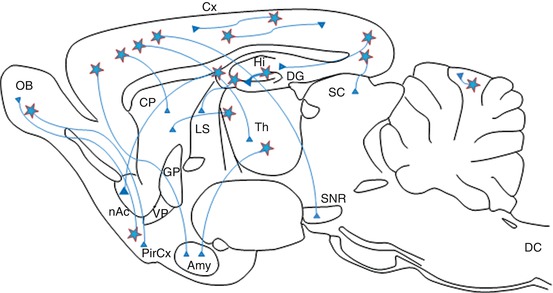
Fig. 3.2
Three-dimensional representation of glutamatergic pathways in the CNS. nAc nucleus accumbens, Amy amygdala, OB olfactory bulb, DC dorsal horn of the spinal cord, CP caudate-putamen, SC superior colliculus, Cx cerebral cortex, PirCx piriform cortex, DG dentate gyrus, GP globus pallidus, Hi hippocampus, LS lateral septum, SNR substantia nigra, Th thalamus, VP ventral pallidum
3.1.1.2 Biosynthesis and Degradation
Glutamate, or glutamic acid, is a nonessential amino acid synthesized by the organism and destined principally for protein synthesis. When utilized as a neurotransmitter, it is synthesized from glutamine within the axon terminal via the action of glutaminase, a mitochondrial enzyme (Fig. 3.6). The glutamate thus formed is stored in synaptic vesicles by an ATP-dependent/sodium-independent active transporter. In the case of glutamate neurons, glial cells recycle GABA and glutamine via the action of glutamine synthetase. Neurons and surrounding glial cells, including astrocytes, take up the glutamate released into the synaptic cleft via specific high-affinity transporters, thus inactivating glutamate’s synaptic actions. After its reuptake, glutamate is converted by glutamine synthase to glutamine, which is then recycled to neurons for the synthesis of glutamate.
3.1.1.3 Glutamate Receptors
Glutamate receptors (see Fig. 3.3) are classified into ionotropic and metabotropic receptors.
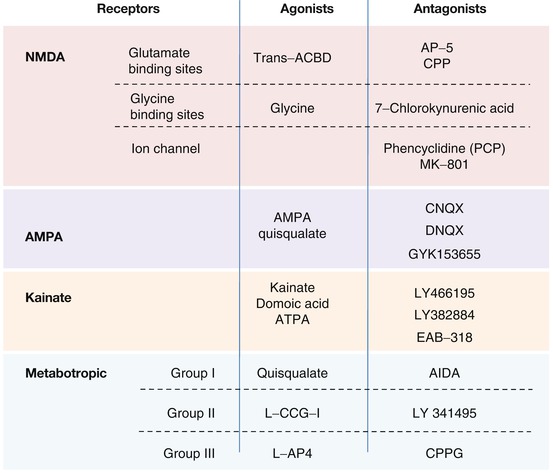
Ionotropic or Ion Channel Receptors
Three types of ionotropic receptor are subclassified according to the exogenous or endogenous agonist substances that they selectively bind:
NMDA receptors are made up of a transmembrane protein complex containing several binding sites and an ion channel permeable to calcium, sodium, and potassium ions. The principal binding site permits the interaction of agonists such as glutamate, NMDA itself, and competitive antagonists such as d-2-amino-5-phosphono-pentanoic acid (d-AP5) and 3-[(+)2-carboxypiperazine- 4-yl] propyl-1-phosphonic acid (CPP). An allosteric site binds glycine and potentiates the action of glutamate. Glycine is indispensable for the activation of the receptor and is blocked competitively by 7-chloro-kynurenate. Two sites are situated within the ion channel: a voltage-dependent cationic site that binds free magnesium (Mg2+) and blocks the calcium channel and a site which binds synthetic compounds such as certain psychotomimetic drugs that act as noncompetitive NMDA receptor antagonists, e.g., phencyclidine (PCP), ketamine, and [5R,10S]-[+]-5-methyl-10,11-dihydro-5H-dibenzo[a,d]cyclohepten-5,10-imine (MK-801, dizocilpine).
AMPA (alpha-amino-3-hydroxy-5-méthyl-4-isoxazole-propionic acid) receptors are ion channels permeable to monovalent cations such as sodium and potassium. Agonists include AMPA itself, quisqualate, and glutamate. There is no specific antagonist for the receptor but certain compounds show a relative selectivity, including 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX) and 6,7-dinitroquinoxaline-2,3-dione (DNQX).
Kainate receptors are permeable to sodium, potassium, and calcium. Agonists include kainic acid, domoic acid, and glutamate. DNQX has been used as a nonselective antagonist.
Metabotropic Receptors
Eight different types of metabotropic glutamate receptors (mGluR1 to mGluR8) are divided into three groups, based on receptor structure and physiological activity. Group I is comprised of receptors mGluR1 and mGluR5. Their activation stimulates the inositol phosphate/diacylglycerol pathway via a pertussis-toxin-sensitive G protein. This induces a rapid increase of intracellular calcium and the activation of protein kinase C. Group II includes mGluR2 and mGluR3 receptors, while group III is made up of GluR4, mGluR6, mGluR7, and mGluR8 receptors. The receptors of these two latter groups inhibit adenylate cyclase via a Gi protein. Selective agonists and antagonists exist for the different receptor groups (see Fig. 3.3).
3.1.1.4 Implication of Glutamate in CNS Disorders
The prolonged application of high concentrations of glutamate has neurotoxic effects resulting from a massive influx of calcium into the neuron – this phenomenon is referred to as excitotoxicity. The neurotoxic property of glutamate and related substances plays an important role in neuronal lesions associated with epilepsy, cerebral ischemia, and traumatic brain injury. Since NMDA antagonist substances can provoke psychotic symptomatologies, it has been suggested that certain psychiatric diseases, such as schizophrenia, may be related to abnormalities in the glutamatergic system.
3.1.1.5 Experimental Tools Influencing the Glutamatergic System
A large number of pharmacological agents are capable of modulating glutamate receptor function (see Fig. 3.3). A selective neurotoxin for the glutamatergic system does not exist. However, it is possible to produce excitotoxic lesions of neurons that express glutamatergic receptors, either directly with glutamate receptor agonists such as kainic acid, ibotenic acid, or NMDA or indirectly, for example, by experimentally provoking an ischemia.
3.1.2 Gamma-Aminobutyric Acid (GABA)
GABA is the major inhibitory neurotransmitter in the nervous system (Fig. 3.4). Its importance is underlined by its deficits associated with epilepsy and anxiety.
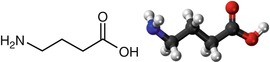
Fig. 3.4
Chemical formula and structure of GABA
3.1.2.1 Brain Localization
GABA is derived from glutamate and is specific to nervous tissue. The cartography of the GABAergic systems (Fig. 3.5) can be revealed using antibodies specific for either GABA or its enzyme of synthesis, glutamate decarboxylase (GAD). GABAergic interneurons are distributed throughout the CNS, including the olfactory bulb, cortex, and hippocampus. Very few structures in the rat brain, such as the ventroposterior thalamic complex, are devoid of these interneurons. The major GABAergic projection systems include the striato-pallido-thalamic, the striato-nigro-thalamic, and the striato-nigro-tectal pathways.
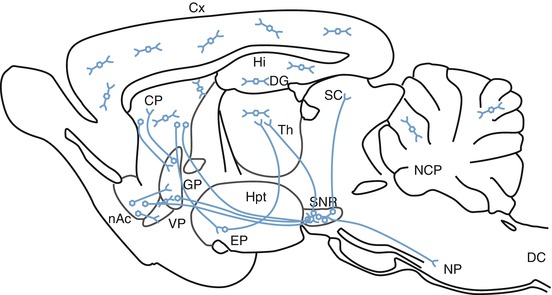
Fig. 3.5
Three-dimensional representation of GABAergic pathways in the CNS (From Epelbaum 1995). nAc nucleus accumbens, CP caudate-putamen, SC superior colliculus, Cx cerebral cortex, DC dorsal horn of the spinal cord, EP entopeduncular nucleus, DG dentate gyrus, GP globus pallidus, Hi hippocampus, Hpt hypothalamus, NCP deep cerebellar nuclei, NP pontine nuclei, SNR substantia nigra reticulata, Th thalamus, VP ventral pallidum
3.1.2.2 Biosynthesis and Degradation (Fig. 3.6)
GAD catalyzes the synthesis of GABA by the decarboxylation of glutamate derived from glutamine. This step requires the presence of the cofactor, pyridoxyl phosphate (PLP or vitamin B6). After its release into the synaptic cleft, GABA is rapidly inactivated by high-affinity reuptake transport systems in both neurons and glia. It is subsequently converted to succinate by two mitochondrial enzymes, GABA transaminase and succinate-semialdehyde dehydrogenase. Succinate thus formed then enters the Krebs cycle. A portion of cellular GABA is not utilized as transmitter – after uptake into mitochondria by membrane exchange, it participates in oxidative metabolism.
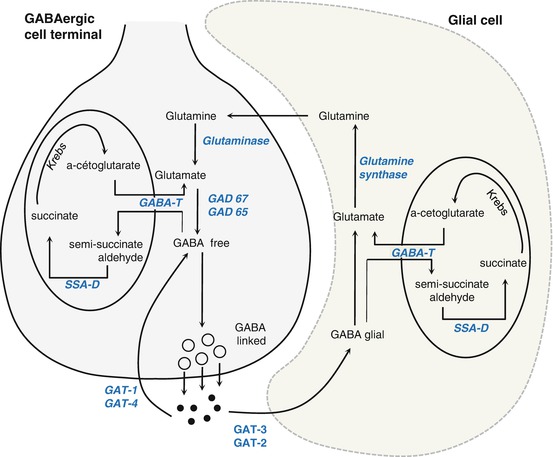
Fig. 3.6
Pathways of biosynthesis and degradation of GABA (From Epelbaum 1995). GABA-T GABA transaminase, GAD glutamate decarboxylase, GAT GABA transporter, SSA-D succinate-semialdehyde dehydrogenase
3.1.2.3 GABA Receptors
Three major types of GABAergic receptors exist: GABAA, GABAB, and GABAC (Fig. 3.7).
GABAA receptors (ionotropic) are coupled to an ion channel specific for chloride ions, the entry of which leads to a hyperpolarization of the cell. This receptor has several different types of binding sites – a site for GABA itself; a benzodiazepine site that potentiates the action of bound GABA through an allosteric effect; a third site that can indiscriminately bind barbiturates, steroids, and alcohol; and a fourth site that binds picrotoxin, a noncompetitive antagonist of GABA. The principal competitive ligands for the GABAA receptor include the agonist muscimol and the antagonist bicuculline (Labrakakis et al. 2003; Bolea et al. 1999; Mirza et al. 2008).
GABAB receptors (metabotropic) are coupled to a Go or Gi protein. The Go protein can be coupled to a calcium channel or a potassium channel, while the Gi protein inhibits adenylate cyclase. The activation of these receptors produces a decrease in calcium currents and as a result reduces the release of neurotransmitters at the nerve terminal level. Their activation also increases potassium ion conductance that causes a hyperpolarization of postsynaptic neurons and a reduction of cAMP leading to reduced transmitter release. Principal ligands for this receptor include the agonist baclofen and the antagonist CGP 56119 (Patenaude et al. 2003).
The GABAC receptor (ionotropic) is coupled to an ion channel specific for chloride ions. Principal ligands include the agonist isoguvacine and the antagonist TPMPA (Yang et al. 2003; Xie et al. 2008).
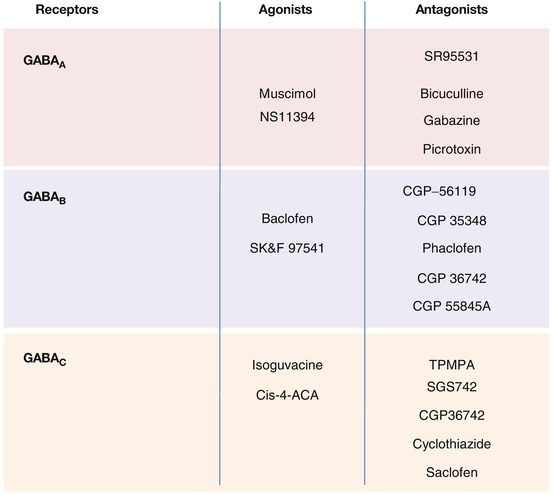
Fig. 3.7
GABA receptors and their agonist and antagonist ligands (Labrakakis et al. 2003; Bolea et al. 1999; Funahashi and Stewart 1998; Shen and Johnson 1997; Kabashima et al. 1997; Patenaude et al. 2003; Mirza et al. 2008; Bon and Galvan 1996; Chebib et al. 2009; Xie et al. 2008; Reis and Duarte 2006; Yang et al. 2003)
3.1.2.4 Implication of GABA in CNS Disorders
A deficit in GABAergic transmission is potentially epileptogenic. Conversely, an increase in GABAergic transmission can have sedative effects as seen with the use of benzodiazepines and barbiturates. An implication of the GABAergic system in certain anxiety disorders seems likely. An alteration in cerebral GABA metabolism has also been associated with neurodegenerative disorders such as Huntington’s disease, in which striatonigral GABAergic neurons are affected.
3.1.2.5 Experimental Tools Influencing the GABAergic System
GABAergic transmission can be modified at different levels: synthesis, degradation, and reuptake. Inhibitors of GAD, such as allylglycine and cycloserine, block GABA synthesis. GABA transaminase, the enzyme of degradation, is inhibited by gamma-vinyl-GABA. Succinate-semialdehyde dehydrogenase is inhibited by sodium valproate. GABA reuptake is inhibited by derivatives of nipecotic acid. Numerous pharmacological agents can modify GABA receptor function (see Fig. 3.7). However, neurotoxins selective for GABAergic neurons are nonexistent.
3.2 The Monoamines
3.2.1 Catecholamines Derived from Tyrosine
3.2.1.1 Noradrenaline
Noradrenaline (Fig. 3.8) has an anatomical and functional duality, as a hormone synthesized and secreted by the adrenal gland and as a neurotransmitter in the central and peripheral nervous systems. Noradrenaline is probably the transmitter implicated in the widest diversity of functions, from the control of blood pressure, attention, and the sleep-wake cycle to cerebral synaptic plasticity.
Fig. 3.8
Chemical formula and structure of noradrenaline
Brain Localization
Histochemical techniques have permitted the localization of noradrenergic neurons by the revelation of noradrenaline and its rate-limiting enzyme of synthesis, dopamine-beta-hydroxylase (DBH). Noradrenergic nerve cell bodies in the CNS are concentrated within the brain stem, in the region bridging the locus coeruleus; the cell groups A4, A5, A6, and A7; the lateral reticular nuclei of the medulla oblongata (group A1); and the nucleus of the tractus solitarius (group A2) (Fig. 3.9; Dahlstrom and Fuxe 1964). Axon bundles from these nuclei disperse widely throughout the CNS to innervate almost every part of the brain. The locus coeruleus has the most diffuse connections in the brain, a single neuron from this nucleus forming as many as 25,000 synapses. Because of this ubiquitous distribution, the locus coeruleus can influence virtually the entire brain.
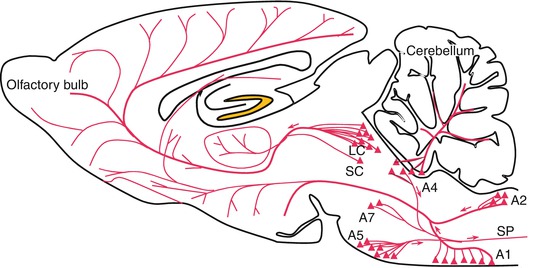
Fig. 3.9
Noradrenergic neurons and their principal projections. Zones A1, A2, A4, A5, and A7 contain noradrenergic neurons. LC locus coeruleus, SC subcoeruleus area, SP spinal cord, LC represents the zone A6
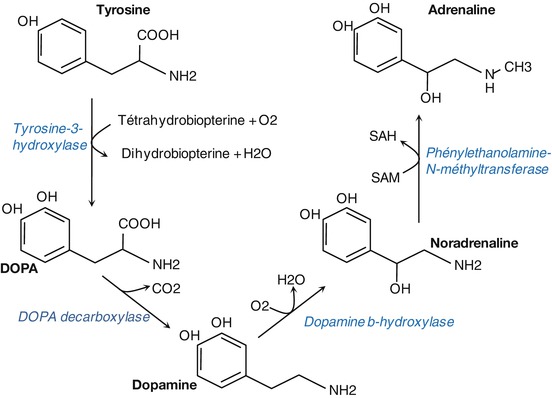
Biosynthesis and Degradation
Noradrenaline is synthesized from the amino acid tyrosine (Fig. 3.10). The active uptake of tyrosine by catecholaminergic neurons is followed by its hydroxylation to l-dihydroxy-phenylalanine (l-Dopa) by tyrosine hydroxylase. l-Dopa is then decarboxylated to form dopamine, which is then taken up and stored within synaptic vesicles where it is converted to noradrenaline by dopamine-beta-hydroxylase.
Released noradrenaline is inactivated by several mechanisms (see Fig. 3.11). A minor portion in the synaptic cleft is transformed directly to normetanephrine by the enzyme catechol-O-methyltransferase (COMT) located in surrounding non-catecholaminergic neurons and glia. The majority of released noradrenaline is taken back up into the nerve terminals by a transmembrane transport system that is specific for noradrenaline. Within the nerve terminal, the recaptured noradrenaline is metabolized to 3,4-dihydroxy-phenyl-glycol-aldehyde by monoamine oxidase A (MAO-A), which converts the functional amine group to an aldehyde. The aldehyde is subsequently transformed to the alcohol and carboxylic acid derivatives.
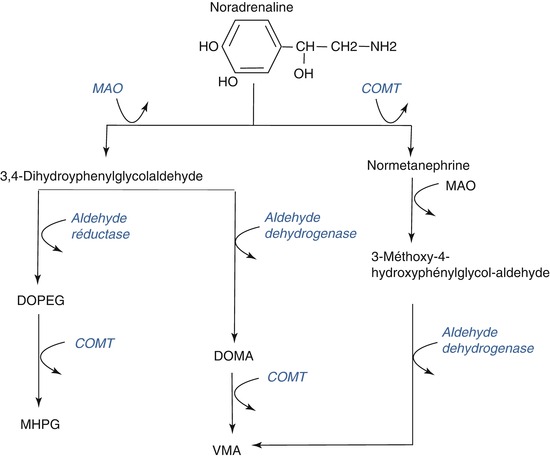
Fig. 3.11
Pathways of noradrenaline degradation. DOPEG dihydroxy-phenyl-ethylene-glycol, MAO monoamine oxidase, COMT catechol-O-methyltransferase, MHPG 3-methoxy-4-hydroxy-phenyl-ethylene-glycol, DOMA dihydroxymandelic acid, VMA vanillylmandelic acid
Receptors
Two types of adrenergic receptors exist, alpha and beta, which are divided into the following subtypes: alpha1a, alpha1b, alpha1d, alpha2a, alpha2b, alpha2c, beta1, beta2, and beta3 (Fig. 3.12; Gether et al. 2002). The stimulation of alpha1 receptors activates the metabolism of phosphoinositides via a Gq protein. These receptors are located for the most part on postsynaptic sites. Alpha2 receptors are coupled to Gi or Go proteins and their activation leads to an inhibition of adenylate cyclase; they are principally localized to presynaptic sites on noradrenergic neurons where they play a role in the negative feedback control of noradrenaline synthesis and release. Alpha2 receptors have also been identified on glutamatergic, serotoninergic, and cholinergic neurons. Beta-adrenergic receptors are localized to postsynaptic sites where their function is excitatory or facilitatory; they are principally coupled to a Gs protein and their stimulation leads to an activation of adenylate cyclase and the formation of cAMP. Presynaptic beta3 receptors play a role in the modulation of catecholamine release (Nasser et al. 2006).
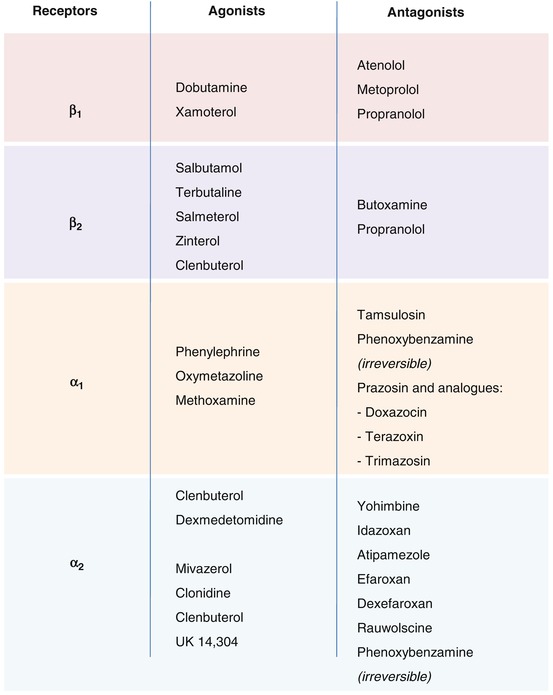
Implication of Noradrenaline in CNS Disorders
In Parkinson’s and Alzheimer’s disease, the locus coeruleus and the medullary noradrenergic cell groups are consistently and profoundly affected (see review by Marien et al. 2004). Cellular loss within the locus coeruleus appears to be more severe in syndromes of dementia. In depression, the implication of the noradrenergic system is based on indirect evidence: reserpine, an inhibitor of vesicular storage of monoamines, can produce depressive symptoms. The majority of clinically effective antidepressant drugs increase the half-life of synaptic noradrenaline by either blocking its reuptake (e.g., imipramine, desipramine, nisoxetine, milnacipran) or blocking its enzymatic degradation (e.g., tranylcypromine).
Experimental Tools Influencing the Noradrenergic System
A number of different neurotoxins exist for the noradrenergic system, for example, N-(2-chloroethyl)-N-ethyl-2-bromobenzylamine (DSP-4) and 6-hydroxydopamine (6-OHDA). However, to improve the selectivity of these neurotoxins, the dopaminergic and serotonergic systems should be protected, respectively, by the concomitant use of specific dopamine and serotonin uptake blockers, such as GBR 12909 and citalopram, respectively. There exist a number of pharmacological tools that are active at different levels of the noradrenergic system, such as amphetamine that acts as a releasing agent and cocaine that blocks noradrenaline reuptake. Caveats for experimental studies using noradrenergic tools are reviewed elsewhere (Marien et al. 2004).
3.2.1.2 Dopamine
In the past, dopamine (Fig. 3.13) was long considered as nothing more than a metabolic intermediate in the synthesis of noradrenaline. Today it is one of the most important neurotransmitters in terms of the diversity and number of functions that it controls and the pathologies in which it is implicated.
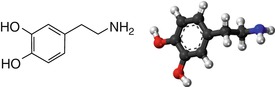
Fig. 3.13
Chemical formula and structure of dopamine
Brain Localization
Histochemical techniques have been used to localize dopamine neurons by the revelation of dopamine and its enzyme of synthesis, tyrosine hydroxylase. The central dopaminergic systems are comprised of several nuclei that constitute anatomically distinct systems (Fig. 3.14):
The mesencephalic system (A8, A9 and A10 nuclei)
The diencephalic system (A11, A12, A13, and A15 nuclei)
The retinal system (A16 and A17 nuclei, not shown in Fig. 3.14)
The mesencephalic system regroups the majority of the dopaminergic cells of the CNS within three juxtaposed structures. The red nucleus, a posterior-lateral extension of the A8 nucleus, and the pars compacta (A9) make up the substantia nigra, adjacent to and with some overlap with the ventral tegmental area cell group (A10). The projections from these cell groups are organized into distinct pathways: the nigrostriatal and mesocorticolimbic systems.
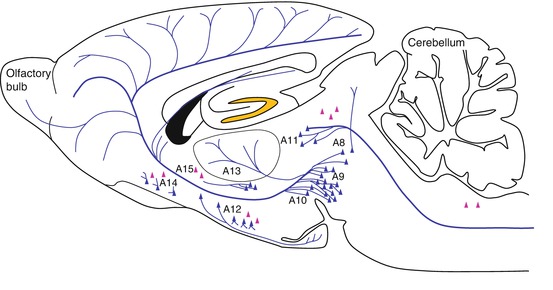
Fig. 3.14
Dopaminergic neurons and their major projections. The blue triangles indicate the localization of dopaminergic neurons. Red triangles indicate groups of l-dopa-positive cells. Blue lines represent dopaminergic fiber projections. A8–A16: dopaminergic cell body groups (Berridge et al. 1997)
The diencephalic system is made up of the diencephalo-spinal system (A11), the incerto-hypothalamic system (A13), the periventricular-preoptic system (A14), the tuberoinfundibular and the tuberohypophysial systems (A12), and neurons in the dorsal preoptic area and ventrolateral hypothalamus (A15). Other dopaminergic neuron groups (not shown in Fig. 3.14) include the periglomerular cells of the olfactory bulb (A16) and neurons in the retina (A17).
Biosynthesis and Degradation
The first steps in the synthesis of dopamine from tyrosine are identical to that of noradrenaline. The active uptake of tyrosine by catecholaminergic neurons is followed by its hydroxylation by tyrosine hydroxylase to l-dopa, which in turn is decarboxylated by DOPA decarboxylase to dopamine. Intraneuronal dopamine is concentrated in storage vesicles that are found in large quantity in the presynaptic axon terminals (Fig. 3.15). Several mechanisms contribute to the inactivation of released dopamine: its recapture into the presynaptic terminals via a specific transmembrane transporter, its uptake into glial cells, and its transformation in the synaptic cleft to 3-methoxytyramine (3-MT) by catechol-O-methyltransferase (COMT). Within the dopamine neuron, dopamine is metabolized by monoamine oxidase B (MAO-B) to dihydroxyphenylacetic acid (DOPAC). DOPAC that passively diffuses out of the neuron is transformed by COMT to homovanillic acid (HVA).
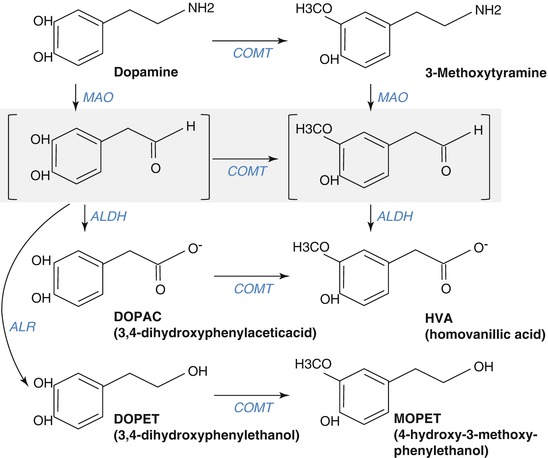
Fig. 3.15




Pathways of dopamine metabolism. The gray zone represents an intermediate reaction step. MAO monoamine oxidase, COMT catechol-O-methyltransferase, ALDH aldehyde dehydrogenase, ALR aldehyde reductase
Related posts:






Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
