Fig. 1.1
Neuroblastic tumor containing abundant neuropil and true rosettes. These tumors characteristically show numerous rosettes, i.e. ependymoblastic (a) and Homer-Wright (b). HE; original magnification (a, b) ×200
Atypical Teratoid Rhabdoid Tumor (AT/ RT)
AT/RT is a rare and highly malignant polyphenotypic neoplasm of uncertain origin that often occurs in the posterior fossa of early childhood. It can be sporadic or can occur in the setting of the rhabdoid tumor predisposition syndrome. This syndrome is characterized by an increased risk to develop malignant rhabdoid tumor in CNS and outside CNS (i.e. kidney, head and neck, soft tissue, heart, mediastinum and liver). AT/RTs are morphologically heterogeneous lesions characterized microscopically by a variable presence of small/embryonal cell, mesenchymal and epithelial features. The most typical, even if inconstant, morphological aspect is the presence of rhabdoid cells with vesicular chromatin, eccentric nuclei, prominent nucleoli, abundant cytoplasm containing inclusion-like mass of filaments.
Pineoblastoma
Pineoblastoma is a rare malignant brain tumor which arises from the pineal gland. It can be sporadic or occur in association with retinoblastoma (ectopic intracranial retinoblastoma) in the setting of the trilateral retinoblastoma syndrome (Blach et al. 1994). Pineoblastoma is a highly cellular neoplasm composed of undifferentiated cells. Pineoblastomas may exhibit histopathological features indicative of photosensory differentiation ranging from immunohistochemical expression of retinal S-antigen to characteristic Flexner–Wintersteiner rosettes and fleurettes reminiscent of retinoblastic differentiation.
Molecular Characterization of Central Nervous System Embryonal Tumors
From the above it may appear that the histopathological diagnosis of the different type of embryonal tumors is easy and that all these malignancies are prognostically and therapeutically equivalent tumors. Actually, it is sometimes difficult to diagnose these tumors with the morphological analysis only, and also not all of these tumors, with an equal stage and treatment, have the same behavior. For these reasons, a molecular approach either for the diagnosis and for the prognostic and therapeutical stratification of these entities, have been advocated.
Medulloblastoma
Many studies have been conducted with the aim of evaluating the genetic basis of medulloblastomas and so obtaining data useful for the diagnosis, the prognosis and the personalization of the therapeutic strategy.
The most common chromosomal aberrations observed in medulloblastomas involve the chromosome 17. Changes in chromosome 17 have been identified in several types of human cancer. In particular, a peculiar chromosomal abnormality called isochromosome 17q (i17q) frequently occurs in some cancers and among these medulloblastomas where has been found in 30–40% of the cases (Pfister et al. 2010). This abnormal version of chromosome 17 combines loss of 17p and gain of 17q. As a result, the chromosome has an extra copy of some genes and is missing copies of other genes. i17p correlates with histological variants and predicts the survival. i17q is more frequently observed in classic and anaplastic/large cell medulloblastoma and has been associated with a poor clinical outcome when compared with that of desmoplastic/nodular medulloblastoma and medulloblastoma with extensive nodularity. This observation suggests that this cytogenetic alteration may contribute to the development of aggressive histotypes (Lamont et al. 2004; Gilbertson and Ellison 2008; Gulino et al. 2008). It has been postulated the existence of tumor suppressor gene(s) on 17p and of oncogene(s) on 17q. In reality, specific genes recurrently mutated in i17q cases have not yet been identified. In this regard it is interestingly to note that TP53 gene, one of the best known and frequently mutated tumor suppressor gene, located at 17p13.1 is not mutated more commonly in medulloblastomas with i17q than those without i17q. On the other hand, the possibility of a inactivation of a specific gene at the 17p breakpoint has been considered unlikely because the breakpoint has a variable localization and occurs in a gene poor chromosomal region (Pfister et al. 2010). Notably, loss of 17p and gain of 17q can also occur independently. Stratification by chromosome 17 data alone demonstrated an unfavorable outcome in patients with isolated 17p loss and suggested a best survival in patients with isolated 17q gain (McCabe et al. 2011).
Alterations in MYC genes have also been described as about 5% of medulloblastomas in mixed cohorts (Lamont et al. 2004; Aldosari et al. 2002). The MYC oncogene family consists of at least three genes: c– (on 8q24), N– (on 2p24), and L– MYC(on 1p32). These proto-oncogenes are among the most powerful activators of tumorigenesis and are frequently overexpressed in several tumors. However, it is still unclear how MYCs promote tumor formation and confer aggressive phenotypes and metastatic potential. It is believed that they act by targeting diverse cellular processes, including cell cycle, proliferation, differentiation, apoptosis, telomere maintenance, cell adhesion and angiogenesis. What is certain is that MYC genes code for proteins that bind to the DNA of other genes and are therefore a transcription factors. They act as activator or, in some cases, repressor, of gene expression (Zitterbart et al. 2011; Takei et al. 2009). The MYC oncoproteins play a central role as regulators of tumorigenesis in numerous different human cancers. Deregulated expression of MYCs is often associated with aggressive, poorly differentiated tumors. High-level amplification (>10-fold) of MYC oncogenes is particularly associated with the large cell/anaplastic phenotype. A moderate MYC oncogenes amplification (>5-fold but <10-fold), particularly c–MYC and N–MYC, is sometimes detected in other histological subtypes and may depend on the presence a large cell/anaplastic component in this medulloblastomas (Eberhart et al. 2004; Stearns et al. 2006; Takei et al. 2009). MYC amplification may also be acquired in recurrent medulloblastomas. Survival analysis showed, independently of histological subtype, a decreasing survival in MYC non-amplified (<5-fold), MYC moderately amplified (<10-fold) and MYC highly amplified (>10-fold) medulloblastomas. Actually, no widely accepted cut-off has been established for defining MYC amplification when, as usual, it is evaluated by FISH method. In a recent report on a large series it has been suggested to considered MYC amplification as clinical relevant when more than 10% of tumor cells exhibit either more than ten signals or innumerable thigh clusters of signals of the respective probe (Pfister et al. 2009). However, lower cut off (MYC copy number >4–5 fold) has been also suggested (Takei et al. 2009) (Fig. 1.2). In any cases it is remarkable that it has been proposed to use the evaluation of the MYC amplification as a molecular stratification factor in the Société Internationale d’Oncologie Pédiatrique en Europe (SIOP Europe) PNET 5 and 6 MB trials. Lastly, some preclinical investigations analyzed the value of MYC expression (particularly c-MYC) in the response of tumors to anticancer therapy. In particular, c-MYC over-expression seems to be associated with enhanced susceptibility of medulloblastoma to radiotherapy- and to etoposide-, doxorubicin-, and cisplatin-induced apoptosis. Consequently, more aggressive treatment approach might be considered in the future to improve the outcome in those patients (von Bueren et al. 2011).
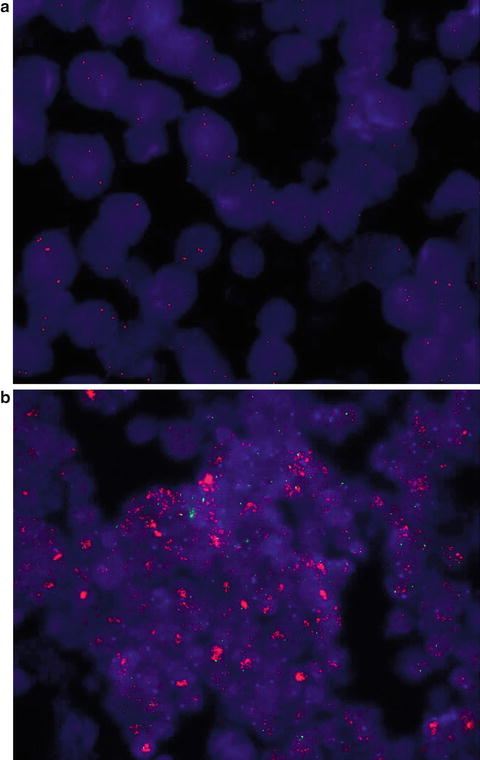
Fig. 1.2
FISH analyses showing not amplified c-MYC gene (less than five red signals for each cells) (a) and amplified c-MYC gene (more than ten signals or innumerable thigh clusters of red signals; green signals correspond to the gene centromere) (b)
The occurrence of medulloblastoma as a possible manifestation of a number of familial syndromes persuaded many authors to investigate whether alterations in the same molecular pathway are implicated in the development of sporadic medulloblastomas.
Particular interest was given to the genetic abnormalities responsible for the type 2 Turcot’s syndrome, in which patients have a predisposition to colon cancer due to mutations in the APC tumor suppressor gene lies on chromosome 5q21. APC protein is a negative regulator of the Wnt/Wingless (Wnt/Wg) pathway. It forms a complex with glycogen synthase kinase 3b and axin and together they control the activity of beta-catenin. Beta-catenin is encoded by the CTNNB1 gene that maps to chromosome 3p22. Beta-catenin binds the actin of the cytoskeleton and is part of a complex of proteins that constitute adherens junctions. Through these skills it is implicated in the transmission the contact inhibition signal that causes cells to stop dividing once the epithelial sheet is complete and in the creation and maintenance of epithelial cell layers by regulating cell growth and adhesion between cells. Sporadic medulloblastomas may show mutation in the Wnt/Wg pathway at different levels: β-catenin, axin and APC genes. Wnt/Wg pathway activation destabilizes the protein complex, upregulating levels of beta-catenin and enhancing its translocation to the nucleus. Here, it cooperate in the regulation of cell cycle progression, apoptosis, and differentiation. Thus, nuclear accumulation of beta-catenin is a marker for physiologic or abnormal Wnt/Wg pathway activation. Mutations of the Wnt/Wg pathway occur in approximately 15% of sporadic medulloblastomas (mainly as a consequence of a mutation in the CTNNB1 gene) and are predicted to cause aberrant pathway activation (Rubin and Rowitch 2002). Children with medulloblastomas that showed a nucleopositive beta-catenin immunophenotype had significantly better overall and event-free survivals than children with tumors that showed either membranous/cytoplasmic beta-catenin immunoreactivity or no immunoreactivity (Rubin and Rowitch 2002; Ellison et al. 2005; Fatet et al. 2009). This finding might be unexpected due to studies of colonic, lung, and hepatocellular carcinomas indicate that nuclear beta-catenin immunoreactivity is associated with both enhanced cell proliferation and an aggressive behavior (Lugli et al. 2007; Martensson et al. 2007). On this regard, we can suppose that multifactorial molecular interactions are involved in the Wnt/Wg pathway in different tumors.
Related posts:
 Oligodendroglial Tumors: Intra-arterial Chemotherapy
The Concept of a Preniche for Localization of Future Metastases
Resection of Brain Tumors: Intraoperative Confocal Microscopy Technology
Oligodendroglial Tumors: Intra-arterial Chemotherapy
The Concept of a Preniche for Localization of Future Metastases
Resection of Brain Tumors: Intraoperative Confocal Microscopy Technology
 Use of Mobile Phones and Brain Cancer Risk in Children?
Use of Mobile Phones and Brain Cancer Risk in Children?
 Metastatic Oligodendroglioma: Diagnosis with Fine-Needle Aspiration Cytology
Metastatic Oligodendroglioma: Diagnosis with Fine-Needle Aspiration Cytology
 Lipoma: An Overview
Lipoma: An Overview
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


