Fig. 17.1
Reward neurocircuitry engaged by chronic drug abuse. AMY, amygdala; DS, dorsal striatum HIPP, hippocampus; NAc, nucleus accumbens; PFC, prefrontal cortex; VP, ventral pallidum; VTA, ventral tegmental area (VTA). Dopaminergic projections from the VTA are depicted by a heavy line. Glutamatergic and γ-aminobutyric acid (GABA)ergic projections, the latter from the NAc to VP, are depicted by thin lines. (For additional information, see (Feltenstein and See 2008; Koob and Volkow 2010; McGinty et al. 2011; Reissner and Kalivas 2013))
A growing appreciation of enduring changes in cellular dynamics within the reward circuitry following chronic drug exposure has fueled the belief that cellular targets of addiction may lie within deficits in executing goal-directed behaviors (Nestler and Aghajanian 1997; Chen et al. 2010; Kalivas and Volkow 2011; Van den Oever et al. 2012) . The appreciation has contributed to a growing shift in the understanding and treatment of addiction as a chronic disease based on cellular pathology following protracted drug abuse (Leshner 1997; Dackis and O’Brien 2005; Chandler et al. 2009; Courtwright 2012) . This shift has in turn fueled study of the relationship between drug-dependent cellular adaptations and addiction-related behaviors. Accordingly, within the last 10 years, changes in glial content, structure, and function have been revealed. The following sections provide an overview of studies from human and preclinical animal models and across levels of analysis from whole brain imaging to cell biology and electrophysiology.
17.2 Astroglial Expression of Receptors and Transporters
In considering the effects of drugs of abuse on glial cells, and on glial contributions to mechanisms of addiction, it is important to appreciate to what degree relevant classes of receptors and transporters are expressed on these cells within the brain reward circuitry. Because the relationship between astrocytes and neurons are the most thoroughly studied in this context, astrocytes will be the predominant glial cell type considered in this section. It has for some time been appreciated that receptors for many transmitter systems are expressed on astrocytes, including glutamate, γ-aminobutyric acid (GABA), serotonin, opioids, purines, and others (Porter and McCarthy 1997; Abbracchio et al. 2006; Shigetomi et al. 2012) . More recently, receptor expression has also been demonstrated for endocannabinoid CB1 receptors, and stimulation of astroglial CB1 receptors by neuronal endocannabinoid release leads to synaptic potentiation via astroglial-derived glutamate transmission (Navarrete and Araque 2008, 2010) . Of particular relevance to this chapter, recent evidence has accumulated showing expression of multiple types of DA receptors in a range of glial cell types in vivo, particularly astrocytes. For example, DA receptors of the subtypes D1, D3–5 (Miyazaki et al. 2004) and D2 (Khan et al. 2001; Shao et al. 2013) have been reported.
Astrocytes also express a wide range of plasma membrane transporters for amino acids, nucleosides, glucose, monocarboxylates, GABA, monoamines, and more (Morgello et al. 1995; Inazu et al. 2003; Pierre and Pellerin 2005; Nagasawa et al. 2007; Fuente-Martin et al. 2012; Kittel-Schneider et al. 2012) . Although astroglial expression of DA transporters has been found in some cases (Takeda et al. 2002; Karakaya et al. 2007), but not others (Dahlin et al. 2007; Kittel-Schneider et al. 2012) , functional uptake of both DA and serotonin in astrocytes has been reported (Kimelberg and Katz 1985; Hirst et al. 1998; Inazu et al. 1999) . Of relevance to this chapter, expression of glutamate transporters on astrocytes has been demonstrated to be of particular importance for protection against excitotoxicity and in maintaining fidelity in synaptic communication. Greater than 90 % of glutamate uptake occurs via high affinity glutamate transporters expressed on astrocytes (Anderson and Swanson 2000; Danbolt 2001) . A deficit in astroglial glutamate uptake has been widely reported following exposure to multiple drugs of abuse, and is discussed in greater detail below.
Functionality of astroglial G protein-linked receptors has also been well described (Bradley and Challiss 2012) . Considerable evidence both in vitro and in vivo indicates that neurotransmitters released from neurons (as well as neuromodulators and hormones) can stimulate astroglial receptors, leading to a Ca2+ signal which triggers release of gliotransmitters and in turn evoked Ca2+ signals in neurons (Bezzi and Volterra 2001; Agulhon et al. 2008; Lu et al. 2009; Zorec et al. 2012) , as well as local and long-distance Ca2+ waves that are propagated through networks of astrocytes via gap junctions (Goldberg et al. 2010) . In vivo, sensory stimulation to a mouse whisker leads to an astroglial Ca2+ response that is dependent upon metabotropic glutamate receptors (mGluRs) (Wang et al. 2006; Halassa and Haydon 2010) .
Signaling from neurons to astrocytes also leads to reciprocal modulation of synaptic and neuronal function by astrocytes themselves (Lu et al. 2009) . This bidirectional nature of communication between neurons and astrocytes and its role in shaping synaptic function has led to the concept of the tripartite synapse (Araque et al. 1999) , which is further supported by electron microscopy and reconstruction studies illustrating the intimate and intricate physical relationship between neurons and astrocytes (Genoud et al. 2006; Witcher et al. 2007; Belanger et al. 2011) .
Because changes in synaptic strength and signaling within the reward neurocircuitry have been heavily implicated in the cellular pathophysiology of addiction , in particular to cocaine (Stuber et al. 2010; Luscher and Malenka 2011) , the morphological and functional relationships between neurons and astrocytes indicate the potential for involvement of astrocytes in mechanisms of drug dependence. Accordingly, the following sections will in more detail discuss evidence for drug-dependent changes in glial cell biology in both human addicts and animal models of addiction, with consideration for how these changes relate to changes in synaptic function.
17.3 Effects of Drug Administration on White Matter Volume and Glial Content
Human brain imaging studies have revealed a great deal of information about neural differences between drug-dependent individuals and healthy, abstinent controls, particularly within regions of the PFC (Goldstein and Volkow 2011) . Brain matter is largely characterized as white and gray matter; white matter is primarily comprised of glial cells and myelinated axons, in contrast to neuronal cell bodies, which are the major constituent of gray matter (Fowler et al. 2007) . Many studies have documented deficits in both volume of both white and gray matter, as well as in metabolic activity in the PFC, of human drug abusers (for review see (Fowler et al. 2007)) .
For example, a number of studies have revealed significant differences in white matter content between drug addicts and healthy controls (Schlaepfer et al. 2006) . Further, while frontal and temporal white matter volume in humans continues to increase until the mid-40s, compared to normal control subjects, cocaine dependent subjects do not exhibit an age-dependent increase in white matter volume (Bartzokis et al. 2002) . Relatedly, deficits in frontal white matter integrity also appear with chronic drug abuse (Lim et al. 2002, 2008) . Integrity is a measure revealed by techniques including diffusion tensor imaging, which quantifies directional diffusion of molecules along a fiber tract, providing an indicator of fiber structure and connectivity (Le Bihan et al. 2001) . White matter integrity across the brain is significantly correlated with treatment outcome for cocaine dependence (Xu et al. 2010) ; relatedly, frontal lobe white matter integrity at the start of treatment is a predictor of alcohol relapse 6 months later (Sorg et al. 2012) . Collectively, these studies indicate that structural deficits in white matter, composed largely of glia , correlate with cortical structural deficits in addicted individuals.
Other studies have reported decreased gray matter measured in a variety of brain regions within cocaine-dependent subjects (Bartzokis et al. 2000; Franklin et al. 2002; Sim et al. 2007) . Prolonged cocaine abuse has been postulated to represent an accelerated aging process with respect to reduction in gray matter volume (Bartzokis et al. 2000; Ersche et al. 2013) . Indeed, the rate of loss of cortical and subcortical gray matter volume in cocaine-dependent subjects is almost twice that of healthy age-matched subjects (Ersche et al. 2013) and accelerated age-dependent loss of gray and white matter has also been reported in chronic alcoholics (Pfefferbaum et al. 1992) .
What is the implication of a prefrontal white matter deficit? Deficient prefrontal cognitive control is a hallmark feature of addiction (Goldstein and Volkow 2002; Kalivas and Volkow 2005; George and Koob 2010) . Impairments of this nature are correlated with increased measures of impulsivity and loss of executive control; for example, decreased frontal and parietal cortex white matter integrity of cocaine-dependent subjects correlates with impaired performs on the Iowa Gambling Task, suggesting that white matter integrity may well be functionally related to functional impairments in decision making (Lane et al. 2010) . However, it is difficult using human studies to dissociate correlation with causation in the relationship between white matter content and integrity with impaired executive function. Furthermore, in all human imaging studies, changes identified between drug-dependent and healthy controls cannot be differentiated between drug-related changes and pre-existing difference which might serve a correlative or causative relationship. As such, preclinical animal models serve a valuable function to allow comparison between naïve baseline and changes which develop with drug experience. Moreover, human studies are confounded by complexity of single drug versus polydrug abusers, length and severity of drug abuse, and other environmental factors and individual differences. These confounds underscore the importance of animal models in understanding the cellular mechanisms responsible for addiction-related behaviors.
In a more limited scope of studies, these principles have been recapitulated in preclinical animal models. For example, diffusion tensor imaging of chronic cocaine treated rats has revealed a significant decrease in white matter integrity of the corpus callosum compared to saline controls; a decrease in myelin-associated protein in the same region was also observed (Narayana et al. 2009) . In a separate study, rats trained to self-administer cocaine demonstrated impairment in a working memory task, which was correlated with decreased counts of both neurons and oligodendrocytes in the PFC (George et al. 2008) . Moreover, drug seeking by rats trained to self-administer cocaine who choose drug seeking despite an adverse consequence (foot shock) was reversed by stimulation of prefrontal cortical activity (Chen et al. 2013) , providing an analog of the hypofrontality observed in human addicts. Future studies will be necessary to further ascertain the relationship between drug abuse, white matter content, and compulsive drug seeking. If then, chronic drug use leads to deficiencies in white and/or gray matter, it becomes important to understand the cellular mechanism(s) by which this may occur, and whether targeting these mechanisms may represent a means of treatment intervention for addiction.
17.4 Effects of Drug Administration on Properties of Astrocytes
17.4.1 Astroglial Inflammation
While functional imaging studies provide important insight into changes at the level of circuitry and anatomy, changes in expression and function of specific proteins provides additional information on glial consequences of drug exposure . A number of studies in preclinical animal models of drug abuse and addiction have demonstrated increased measures for astrocyte activation. For example, increased glial fibrillary acidic protein (GFAP) expression within the reward neurocircuitry has been observed following exposure to cocaine (Fattore et al. 2002; Bowers and Kalivas 2003) , methamphetamine (Guilarte et al. 2003; Friend and Keefe 2013) , morphine (Beitner-Johnson et al. 1993; Song and Zhao 2001) , and alcohol (Fletcher and Shain 1993; Goodlett et al. 1993; Franke 1995; Tagliaferro et al. 2002; Vongvatcharanon et al. 2010) . Increased GFAP expression is a hallmark feature of inflammation (or activation) of astrocytes (Pekny and Nilsson 2005; Sofroniew 2009; Sofroniew and Vinters 2010) and hence may reflect enduring cellular inflammation initiated by drug exposure. In some cases, however, long-term alcohol exposure has also been associated with a decline in GFAP expression and astrocyte complexity (Franke 1995; Rintala et al. 2001) . Detailed histological analysis on postmortem alcoholic human brains have revealed enlarged cell bodies but also patch-like losses in GFAP-positive cells (Cullen and Halliday 1994; Miguel-Hidalgo 2005) , indicating a complexity of cellular effects. Nonetheless, many studies have revealed that exposure to drugs of abuse leads to glial inflammation and activation of innate immune response (for reviews see (Watkins et al. 2005, 2009; Crews et al. 2011)) .
17.4.2 Blood-Brain Barrier Integrity
Psychostimulant abuse has also been well documented to lead to breakdown of the blood brain barrier (BBB) (Kousik et al. 2012) . The BBB is a physical barrier to the central nervous system (CNS) formed by brain endothelial cells interacting with pericytes, basement membrane of the vasculature, and astrocytes (Banerjee and Bhat 2007; Abbott et al. 2010; Krueger and Bechmann 2010; Kousik et al. 2012) . The tight association of astrocyte end-feet with vasculature is a critical component, and astroglial inflammation can lead to perturbation of BBB integrity. Permeability of the BBB for any reason leads to vulnerability to CNS exposure to a variety of chemical and biological insults, from which it is normally protected. A good example is the fact that mounting evidence indicates increased vulnerability of the nervous system to HIV infection in psychostimulant abusers (Rippeth et al. 2004; Buch et al. 2012) . HIV infection within the brain in turn leads to an increased incidence of neuro-AIDS, a condition characterized by generalized neurologic deficits secondary to HIV infection (Buch et al. 2012; Hauser et al. 2012) . The abuse of psychstimulants such as cocaine and methamphetamine leads to glial inflammation and permeability of the BBB, enhanced vulnerability to HIV infection and other opportunistic viruses and pathogens.
17.4.3 Astroglial Glutamate Transport: A Critical Mediator of Drug Seeking
Among the most widely reported astroglial changes following self-administration of drugs of abuse is downregulation of the high affinity glutamate transporter EAAT2/GLT-1 (Reissner and Kalivas 2010) . Downregulation of GLT-1 is frequently associated with activated astrocytes, particularly following injury (Binns et al. 2005; Pekny and Nilsson 2005; Cata et al. 2006; Tawfik et al. 2008) . GLT-1 is downregulated in the NAc following self-administration and withdrawal from cocaine (Knackstedt et al. 2010; Fischer-Smith et al. 2012) , nicotine (Gipson et al. 2013) , and heroin (Shen et al. personal communication). Results following alcohol exposure are more complex; however, in one study 24 h after 7 days alcohol exposure, higher basal extracellular glutamate levels and corresponding decrease in glutamate uptake was reported in the NAc (Melendez et al. 2005) , which is consistent with other studies reporting chronic increases in basal extracellular glutamate levels (Mann et al. 2008) . Moreover, chronic self-administration in alcohol-preferring rats results in decreased GLT-1 protein levels in the NAc; treatment with ceftriaxone reversed this decreased, as well as measures of alcohol seeking in alcohol-preferring rats (Qrunfleh et al. 2013; Sari et al. 2013) .
GLT-1/EAAT2 is a member of the EAAT family of high affinity glutamate transporters (Danbolt 2001), and rapid glutamate uptake is important for the tight regulation of synaptic glutamate concentrations and the prevention of synaptic excitotoxicity (Danbolt 2001; Jiang and Amara 2011) . In addition, impaired clearance of glutamate from the synaptic cleft can impact decay currents of ionotropic glutamate receptors, indicating that at sub-excitotoxic levels, changes in glutamate concentrations resulting from altered transporter function can shape neuronal currents (Tzingounis and Wadiche 2007) . Importantly, restored expression of GLT-1 is associated with decreased measures of cocaine seeking (Baker et al. 2003; Knackstedt et al. 2010; Reissner et al. 2013) .
Interestingly, besides following drug abuse, EAAT2/GLT-1 levels and transporter-dependent glutamate uptake are also decreased in aged rodent brains (Wheeler and Ondo 1986; Najlerahim et al. 1990; Saransaari and Oja 1995; Vatassery et al. 1998; Potier et al. 2010) as well as in aged brains in a transgenic mouse model of Huntington’s Disease (Behrens et al. 2002) and in postmortem brains from Alzheimer’s Disease (AD) patients (Masliah et al. 1996; Jacob et al. 2007; Simpson et al. 2010; Woltjer et al. 2010) . Moreover, heterozygous-deficient GLT-1 (+/−) mice crossed with a double transgenic AD model mouse demonstrated an earlier onset of memory deficits compared with the AD model mouse with a wild-type GLT-1 genotype (Mookherjee et al. 2011) . These findings collectively indicate that impaired GLT-1 expression and function may contribute to age-dependent synaptic and cognitive impairment, and is supportive of the idea that cocaine-dependent changes may accelerate the aging process (Ersche et al. 2013) .
While synaptic glutamate levels are predominantly controlled by GLT-1, basal extracellular glutamate levels are correspondingly largely controlled by the action of the cystine-glutamate exchanger, system xc− (Baker et al. 2002a, 2002b) . System xc− is composed of a small, catalytic subunit, xCT, and a large subunit, NF2 (Lim and Donaldson 2011; Lewerenz et al. 2013) . While some neuronal expression of system xc− has been reported, it is predominantly astroglial. Hence, extracellular glutamate levels are controlled by release of glutamate via system xc− , and uptake by EAATs (largely GLT-1 in adult rodents). Interestingly, while expression of both xCT and GLT-1 are decreased following cocaine self-administration and extinction training (Knackstedt et al. 2010) , net basal extracellular glutamate levels are decreased, indicating that expression of xc− may be the more dominant toward controlling basal extracellular levels measured via microdialysis (Baker et al. 2003) . This has led to a model in which basal levels are decreased by suppressed xCT, but synaptic levels are increased following glutamate release, due to the compromised synaptic uptake cause by impaired GLT-1 function and expression (Fig. 17.2) (McFarland et al. 2003) .
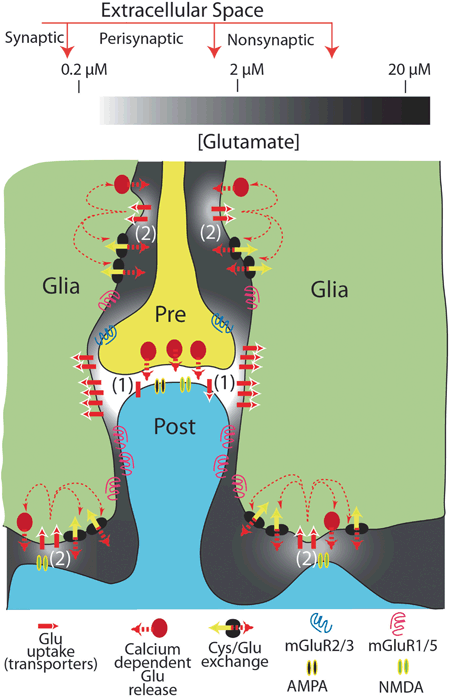
Fig. 17.2
Cystine/glutamate exchange and glutamate uptake regulate synaptic and extrasynaptic glutamate levels. Both the glutamate transporter GLT-1 and glutamate exchanger catalytic subunit xCT are reduced following chronic self-administration and extinction from cocaine. Ca2+-dependent glutamate release from astrocytes is also documented, although cystine/glutamate (Cys/Glu)exchange is the primary source (Baker et al. 2002a; Haydon et al. 2009). Synaptic glutamate levels (1) are tightly controlled within the sub-micromolar range at rest (during synaptic transmission glutamate can raise to ~ 1.1 mM), whereas extrasynaptic levels (2) can approach the 20 µM range. Reduced expression of xCT will lead to decreased basal extrasynaptic levels, and hence reduced tone of presynaptic inhibitory mGluR2/3 receptors. This leads to increased glutamate release (and hence, synaptic levels) within the NAc during cocaine seeking, which is exacerbated by reduced GLT-1 expression. Inotropic glutamate, AMPA (α-amino-3-hydroxy-5-methyl-isoxazole propionate) and NMDA, receptors are shown. (Figure reproduced with permission from (Moussawi et al. 2011))
A consequent dysregulation of synaptic and extrasynaptic glutamate levels following suppression of xCT and GLT-1 has considerable consequences on cellular function (Kalivas 2009) . Decreased basal levels of extracellular glutamate (due to decreased xCT) lead to decreased tone on presynaptic inhibitory mGluR2/3 receptors, which results in increased glutamatergic transmission. Activity of glutamatergic PFC innervation of medium spiny neurons of the NAc is a driving force of reinstatement to numerous drugs of abuse, and pharmacological agonism of these presynaptic mGluR2/3 receptors (or antagonism of postsynaptic excitatory glutamate receptors) can block reinstatement (Moussawi and Kalivas 2010; Reissner and Kalivas 2010) . Hence, the decreased expression of xCT and GLT-1 leads to a combinatorial effect on the cellular controls designed to regulate synaptic input to the NAc.
Extracellular glutamate levels appear not only to mediate tone on neuronal receptors, but can also stimulate Ca2+ oscillations in astrocytes via stimulation of mGluR receptors, as discussed in the preceding section. This Ca2+-dependent glutamate transmission from astrocytes leads to N-methyl D-aspartate receptors (NMDARs)-mediated slow inward currents (SICs) on medium spiny neurons in the NAc (D’Ascenzo et al. 2007) . What is the behavioral consequence of changes in glutamate transmission from astrocytes? One clue comes from transgenic mice engineered to express a dominant negative vesicle protein in astrocytes; these mice demonstrate impaired cocaine-induced reinstatement of conditioned place preference, as well as cue-induced reinstatement in a self-administration paradigm (Turner et al. 2013) . These findings are consistent with a model in which glutamatergic gliotransmission may mediate the cellular pathology of addiction. However, more studies will be required to fully understand the consequences of impairments of gliotransmission following drug abuse.
17.5 Beyond Astrocytes: Effects of Drug Administration on Oligodendrocytes and Microglia
In addition to cellular adaptations within astrocytes, drug-dependent changes in oligodendrocytes and microglia have been reported. Withdrawal from chronic cocaine administration is associated with decreased expression of white matter proteins in the NAc including myelin basic protein, proteolipid protein (PLP), myelin oligodendrocyte glycoprotein, and myelin associated glycoprotein, which is prevented by treatment with the β-lactam antibiotic ceftriaxone (Kovalevich et al. 2012) . As mentioned above, ceftriaxone is a known inducer of GLT-1 expression (Rothstein et al. 2005) , and is effective at preventing reinstatement of cocaine (Sari et al. 2009; Knackstedt et al. 2010; Fischer et al. 2013) . Interestingly, postmortem gene expression profiling in NAc tissue from human addicts has revealed a decrease in mRNA coding for myelin-associated proteins, including myelin basic protein and PLP1 (Albertson et al. 2004; Bannon et al. 2005) . In a separate study, a decrease in PLP1 was confirmed by in situ hybridization in the ventral caudate, putamen, and internal capsule of postmortem tissue from human cocaine abusers (Kristiansen et al. 2009) .
As stated in the preceding section, it is well documented that activation of glial cells occurs in response to opioid exposure. However, in contrast to the analgesic effects of opioids that are mediated through opioid receptors, activation of glial cells appears to occur via toll-like receptors (TLRs) and mediate nonanalgesic effects of opioids (Hutchinson et al. 2011) . TLRs are microbe receptors expressed on immune cells, named for homology to the Drosophila Toll protein (Anderson et al. 1985; Lemaitre et al. 1996) . The signaling of TLRs is a critical component in activating an immune response (Hanke and Kielian 2011) . Within the CNS, TLRs are expressed predominantly on microglia, but are found on activated astrocytes as well, and on oligodendrocytes and neurons to a lesser degree (Hanke and Kielian 2011) . TLR4 serves as the receptor for lipopolysaccharide, which has allowed for elucidation of a complex downstream signaling cascade leading to increased transcriptional regulation by nuclear factor kappa-light-chain-enhancer of activated B cells (Kawai and Akira 2006) . Activation of the TLR4 receptor in response to opioid exposure in vivo opposes the analgesic effects and is responsible for opioid-induced hyperalgesia, or hypersensitivity to pain that often occurs after long term opioid use and abuse and in fact oppose the analgesic effects mediated through opioid receptor (Ossipov et al. 2005; Buchanan et al. 2010) . Effects of opioid versus TLR actions can be dissociated using (−) and (+) stereoisomers of opioid agonists and antagonists; while TLRs are nonstereoselective, opioid receptors only bind to (−)-isomers. Genetic deletion or pharmacological inhibition of TLR4 receptors with (+)-naloxone impair opioid-induced CPP and reduce opioid self-administration (Hutchinson et al. 2012) .
17.6 Modulation of Neuronal Cell Biology by Glial-derived Factors
17.6.1 GDNF, A Negative Regulator of Drug Reward
In the preceding section, we introduced the concept of glutamatergic gliotransmission, and how the regulation of glutamate release and uptake by astrocytes may contribute to disruptions in glutamate homeostasis and drug-induced cellular pathologies (Cali et al. 2009) . Importantly, glial cells and in particular astrocytes secrete many proteins and molecules in addition to glutamate which may also be adaptive or inflammatory in response to a protracted drug history. For example, glial cell line-derived neurotrophic factor (GDNF) is a member of the transforming growth factor-βfamily of growth factors that, despite its glio-centric name, is expressed in a variety of cell types including astrocytes, microglia , and neurons (Schaar et al. 1993; Pochon et al. 1997) . GDNF is predominantly expressed within the basal forebrain, substantia nigra, and striatum, and was initially isolated on the basis of its ability as a component of astrocytes to selectively protect DA neurons of the substantia nigra in culture (O’Malley et al. 1992; Schaar et al. 1993; Duarte et al. 2012) . A number of studies collectively indicate that GDNF action opposes drug taking, as well as the effects of chronic exposure on seeking of multiple drugs of abuse (Pierce and Bari 2001; Carnicella and Ron 2009; Ghitza et al. 2010) . However, manipulation of GDNF expression and activity in the VTA promotes the incubation of cocaine craving (Lu et al. 2009) but not heroin craving (Airavaara et al. 2011) , indicating the different roles may exist in difference brain regions and at different times following drug exposure.
17.6.2 Gliotransmitters: Beyond Glutamate
D-serine is a naturally occurring amino acid transmitter mainly released by astrocytes (and neurons in some brain regions), and is a co-agonist for NMDA receptors (Ben Achour and Pascual 2012; Radzishevsky et al. 2013) . Stimulation of NMDARs by D-serine is necessary for hippocampal NMDA-dependent long-term potentiation. Relatedly, D-cycloserine (DCS) is a broad spectrum antibiotic and partial agonist at the glycine site of NMDA receptors, with functions as a cognitive enhancer and anticonvulsant (Ohno and Watanabe 1996; Wlaz et al. 1996; Myers and Carlezon 2012) , and is under investigation as a treatment for substance use disorder (Olive et al. 2012) .
A growing body of evidence suggests that administration of either D-serine or DCS may reduce drug seeking. Reduced D-Serine levels are observed in the NAc following cocaine administration (Curcio et al. 2013) . Further, perfusion of D-Serine in the bath of NAc core slice electrophysiology restored the ability to induce long-term potentiation and depression. Relatedly, microinjection of D-Serine in the NAc blocked behavioral sensitization to cocaine . D-Serine administered systemically prior to or immediately after extinction training sessions significantly reduces the reinstatement of cocaine seeking (Kelamangalath and Wagner 2010) . Similarly, systemic administration of D-serine or DCS blocks expression of cocaine conditioned place preference (CPP) (Yang et al. 2013). DCS facilitates extinction of cocaine CPP (Botreau et al. 2006) . Moreover, the effect of DCS can be long-lasting and reduce reinstatement of the CPP behavior (Paolone et al. 2009; Thanos et al. 2009) . A Phase 2, “D-Serine for Cocaine Dependence Pilot” clinical study is currently underway (http://www.clinical trials.gov NCT01715051).
Besides glutamate and D-Serine, ATP is an additional gliotransmitter released by astrocytes. Extracellular adenosine is largely derived from astroglial ATP, acting on neuronal adenosine receptors. Evidence exists for a role of adenosine receptors, in particular A2A receptors, in the rewarding effects of drugs of abuse, and in reinstatement to drug seeking (Soria et al. 2006; Brown et al. 2009; O’Neill et al. 2012) .
17.7 Glial Cells: An Emerging Pharmacotherapeutic Target for Addiction
The study described above illustrates the potential for astrocyte-derived D-serine as pharmacotherapy for substance abuse disorder. D-serine is one of a number of examples of therapies based in glial cell biology, and adaptions in neuron-glial signaling (Cooper et al. 2012) . For example, as stated previously, at least three compounds known to induce expression of GLT-1 are also under investigation as addiction therapies. N-acetylcysteine, ceftriaxone, and propentofylline are all compounds of separate classes which upregulate GLT-1, and suppress reinstatement to cocaine (Baker et al. 2003; Sari et al. 2009; Knackstedt et al. 2010; Reissner et al. 2014) . Human clinical trials indicate safety and efficacy of N-acetylcysteine against drug use for nicotine, cocaine, and cannabis (LaRowe et al. 2007, 2013; Knackstedt et al. 2009; Gray et al. 2012; Berk et al. 2013) . Further, the restored expression of GLT-1 is necessary in order for propentofylline to effectively block reinstatement to cocaine (Reissner et al. 2014) . Thus, while the field of gliobiology in drug abuse is in a relatively early stage, credible evidence already exists to support the hypothesis that targeting glial physiology affects neuronal cell biology and drug craving.
17.8 Conclusions and Perspectives
There is no question that both acute and chronic exposure to drugs of abuse leads to changes in astrocytes, oligodendrocytes , and microglia, within regions heavily implicated in reward neurocircuitry. However, our understanding of the effects of drug-dependent changes in glial structure and activity is at an early stage. Important questions remain to be addressed, such as: what are the relationships between changes in expression of myelin-related genes, deficits in white matter volume and integrity, and compulsive drug seeking? What are the relationships between glial inflammation and changes in white matter content, or between drug-dependent changes in astrocyte physiology and neuronal synaptic communication? These questions will be central toward future progress in understanding and treating addiction as a brain disease.
References
Abbott NJ, Patabendige AA, Dolman DE, Yusof SR, Begley DJ (2010) Structure and function of the blood-brain barrier. Neurobiol Dis 37:13–25PubMed
Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Knight GE, Fumagalli M, Gachet C, Jacobson KA, Weisman GA (2006) International union of pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev 58:281–341PubMedPubMedCentral
Agulhon C, Petravicz J, McMullen AB, Sweger EJ, Minton SK, Taves SR, Casper KB, Fiacco TA, McCarthy KD (2008) What is the role of astrocyte calcium in neurophysiology? Neuron 59:932–946PubMedPubMedCentral
Airavaara M, Pickens CL, Stern AL, Wihbey KA, Harvey BK, Bossert JM, Liu QR, Hoffer BJ, Shaham Y (2011) Endogenous GDNF in ventral tegmental area and nucleus accumbens does not play a role in the incubation of heroin craving. Addict Biol 16:261–272PubMedPubMedCentral
Albertson DN, Pruetz B, Schmidt CJ, Kuhn DM, Kapatos G, Bannon MJ (2004) Gene expression profile of the nucleus accumbens of human cocaine abusers: evidence for dysregulation of myelin. J Neurochem 88:1211–1219PubMedPubMedCentral
Anderson CM, Swanson RA (2000) Astrocyte glutamate transport: review of properties, regulation, and physiological functions. Glia 32:1–14PubMed
Anderson KV, Bokla L, Nusslein-Volhard C (1985) Establishment of dorsal-ventral polarity in the drosophila embryo: the induction of polarity by the toll gene product. Cell 42:791–798PubMed
Araque A, Parpura V, Sanzgiri RP, Haydon PG (1999) Tripartite synapses: glia, the unacknowledged partner. Trends Neurosci 22:208–215PubMed
Baker DA, Shen H, Kalivas PW (2002a) Cystine/glutamate exchange serves as the source for extracellular glutamate: modifications by repeated cocaine administration. Amino acids 23:161–162
Baker DA, Xi ZX, Shen H, Swanson CJ, Kalivas PW (2002b) The origin and neuronal function of in vivo nonsynaptic glutamate. J Neurosci 22:9134–9141
Baker DA, McFarland K, Lake RW, Shen H, Tang XC, Toda S, Kalivas PW (2003) Neuroadaptations in cystine-glutamate exchange underlie cocaine relapse. Nat Neurosci 6:743–749PubMed
Related posts:
 General Pathophysiology of Neuroglia: Neurological and Psychiatric Disorders as Gliopathies
General Pathophysiology of Neuroglia: Neurological and Psychiatric Disorders as Gliopathies
 Ionic Signaling in Physiology and Pathophysiology of Astroglia
Ionic Signaling in Physiology and Pathophysiology of Astroglia
 Enteric Glial Cells: Implications in Gut Pathology
Enteric Glial Cells: Implications in Gut Pathology
 Astroglial Connexins as Elements of Sleep-Wake Cycle Regulation and Dysfunction
Astroglial Connexins as Elements of Sleep-Wake Cycle Regulation and Dysfunction
 Novel Therapeutic Approaches to Malignant Gliomas
Novel Therapeutic Approaches to Malignant Gliomas
 Neurodegeneration and Neuroglia: Emphasis on Astroglia in Alzheimer’s Disease
Neurodegeneration and Neuroglia: Emphasis on Astroglia in Alzheimer’s Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


