Fig. 10.1
Increase in the number of primary research reports in psychiatric or behavioral epigenetics. This literature search was done in PubMed by searching for publications in the specified year that met the following criteria: epigenetic or DNA methylation or histone or chromatin or miRNA or ncRNA and psychopathology or psychiatric or behavior
Overview of Epigenetics
All somatic cells within an organism contain the same DNA sequence, but each type of cell expresses different genes based on its specialized function. This is because each cell type has a unique epigenetic pattern that dictates the timing and magnitude of gene expression by restricting areas of the genome available for transcription. Once established, epigenetic patterns are generally maintained through mitosis (Bonasio, Tu, & Reinberg, 2010). However, some aspects of the epigenome are receptive to environmental conditions that change during an organism’s lifetime. Thus, aspects of the epigenome can respond to changes in the environment by altering gene expression (Feil & Fraga, 2011).
The definition of epigenetics has changed since Conrad Waddington first coined it in 1942 as “the branch of biology which studies the causal interactions between genes and their products which bring the phenotype into being” (Waddington, 1942). The word epigenetics comes from the Greek “epi-,” meaning “above” genetics. Since that time, the definition of epigenetics continues to evolve. The field currently encompasses the study of covalent and non-covalent modifications of DNA, its associated proteins, and RNA transcripts that regulate gene expression. Epigenetic mechanisms can induce changes in gene expression independent of variation in the DNA sequence and are maintained through each round of cell division. They respond to changes in the environment, are potentially reversible, and thus can be targeted for disease therapies (Feinberg, 2007).
There is approximately 2–3 meters of DNA in the nucleus of a human cell (Campbell, Mitchell, & Reece, 1997). To protect and organize the DNA, it is wrapped around packaging proteins (histones), and the unit is called a nucleosome (Fig. 10.2) (Annunziato, 2008). When assembled as a group, nucleosomes make up chromatin, which prevents DNA damage while regulating DNA replication and gene expression. Chromatin is classified as either heterochromatin or euchromatin based on its conformation (Fig. 10.3). Heterochromatin promotes DNA silencing because a close compaction of the nucleosomes restricts access of the transcriptional machinery to the DNA. Euchromatin promotes active transcription of DNA as its nucleosomes are not as tightly packed (Li, Carey, & Workman, 2007).
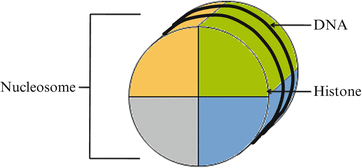
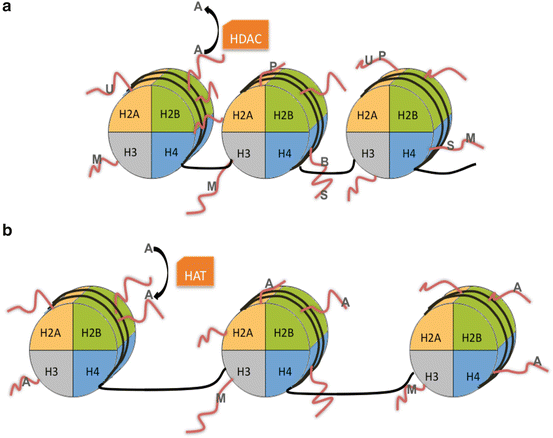
Fig. 10.2
A nucleosome is the unit of DNA wrapped around a group of core histones
Fig. 10.3
Heterochromatin (a) is a densely packed conformation of DNA that resists active transcription. Heterochromatin formation is associated with specific histone modifications (shown in gray) including biotinylation (B) of H4; methylation (M) of H1, H3, or H4; phosphorylation (P) of H2A; sumoylation (S) of H4; and ubiquitylation (U) of H2A. Histone deacetylases (HDACs) remove acetyl (A) groups, which are associated with active transcription. Histone acetyltransferases (HAT) transfer acetyl groups onto the histone tails when chromatin relaxes to become euchromatin (b). Histone modifications that correlate with euchromatin formation are acetylation (A) and methylation (M) of H3
Both core (H2A, H2B, H3, H4) and linker (H1 and H5) histones can be modified through methylation, acetylation, phosphorylation, ubiquitination, sumoylation, and biotinylation as detailed in Table 10.1. Novel histone marks and posttranslational modifications are continually being discovered. Each modification has a specific function that can influence transcription, chromatin conformation, and cofactor recruitment. For example, histone acetylation typically promotes gene transcription while histone methylation can promote or repress transcription based on where it is occurring (Bannister & Kouzarides, 2011).
Table 10.1
Types and functions of histone modifications
Modification | Histone | Location | Function | References |
|---|---|---|---|---|
Acetylation | H2A | Lys5 | Transcriptional activation | Schiltz et al. (1999) |
H2B | Lys5 | Transcriptional activation | Schiltz et al. (1999) | |
Lys12 | Transcriptional activation | Schiltz et al. (1999) | ||
Lys15 | Transcriptional activation | Schiltz et al. (1999) | ||
Lys20 | Transcriptional activation | Schiltz et al. (1999) | ||
H3 | Lys9 | Transcriptional activation | Kurdistani, Tavazoie, and Grunstein (2004) | |
Lys14 | Transcriptional activation | Kurdistani et al. (2004) | ||
H4 | Lys5 | Transcriptional activation | Schiltz et al. (1999) | |
Lys8 | Transcriptional activation | Schiltz et al. (1999) | ||
Lys12 | Transcriptional activation | Schiltz et al. (1999) | ||
Methylation | H1 | Lys26 | Transcriptional suppression | Kuzmichev, Jenuwein, Tempst, and Reinberg (2004) |
H3 | Lys4 | Transcriptional activation | Martin and Zhang (2005) | |
Lys9 | Heterochromatin formation | Martin and Zhang (2005) | ||
Lys27 | X-inactivation, genomic imprinting, transcriptional suppression | Martin and Zhang (2005) | ||
Lys36 | Transcriptional suppression | Martin and Zhang (2005) | ||
Lys79 | Transcriptional suppression | Martin and Zhang (2005) | ||
Arg8 | Transcriptional suppression | Wang, Pal, and Sif (2008) | ||
H4 | Lys20 | Heterochromatin formation | Martin and Zhang (2005) | |
Arg3 | Transcriptional suppression | Wang et al. (2008) | ||
Phosphorylation | H2A.X | Ser139 | DNA double strand repair | Foster and Downs (2005) |
Sumoylation | H4 | Transcriptional suppression | Shiio and Eisenman (2003) | |
Ubiquitylation | H2A | Lys119 | Transcriptional suppression | Zhou et al. (2008) |
Biotinylation | H4 | Lys12 | Transcriptional suppression | Camporeale, Oommen, Griffin, Sarath, and Zempleni (2007) |
Histone modifications often correlate with changes in methylation of DNA. In adults, this reaction occurs when a DNA methyltransferase (DNMT) catalyzes the transfer of a methyl group from a donor, such as S-adenosylmethionine (SAM), to the 5′ carbon of cytosine in the context of a cytosine phosphodiester guanine (CpG) dinucleotide, commonly called a CpG site (Fig. 10.4). DNA methylation regulates gene expression by influencing the recruitment and binding of regulatory proteins to DNA; typically, an increase in methylation at gene promoter regions is correlated with a decrease in expression of that gene (Bonasio et al., 2010).
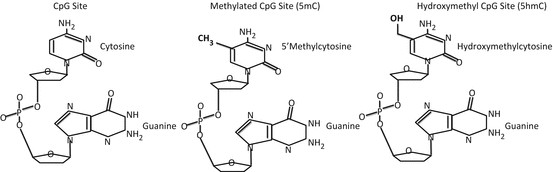
Fig. 10.4
When a cytosine and guanine are adjacent to one another, the cytosine can be methylated in the 5′ position, creating 5′ methylcytosine. TET enzymes can add a hydroxyl group creating hydroxymethylcytosine
However, DNA methylation patterns are not constant across a gene; they vary based on the genetic architecture of a region. For example, the majority of DNA methylation across the genome occurs in specific groups of repetitive sequences (i.e., LINEs and SINEs) (Gu, Wang, Nekrutenko, & Li, 2000). These repetitive elements are often heavily methylated, and loss of DNA methylation in these regions has been associated with cancer (Ehrlich, 2002).
In transcriptionally active genes, DNA methylation patterns relate to specific regulatory functions. For example, CpG sites are overrepresented in the promoter region of many genes, and if they cluster in sufficient density, the region is called a CpG island (Weber et al., 2007). Islands typically have very low methylation levels, but the regions near islands, called CpG shores, exhibit lower cytosine density and are more likely to harbor tissue-specific and disease-specific methylation patterns (Irizarry et al., 2009). Carrying the analogy forward, regions beyond shores can be termed as CpG shelves (Irizarry et al., 2009). While these studies focus on the promoter and 5′ region of the gene, intragenic DNA methylation patterns are also important to regulate alternative promoters and enhancers that define tissue specificity in a variety of alternative transcripts (Maunakea et al., 2010).
The majority of studies to date focus on 5-methylcytosine (5mC), but CpG sites can be further modified through hydroxylation to produce 5-hydroxymethylcytosine (5hmC; Fig. 10.4) (Munzel, Globisch, & Carell, 2011), which plays an intermediate role in oxidative demethylation pathways (Guo, Su, Zhong, Ming, & Song, 2011). Across the genome, 5hmC is associated with increased transcription and highly involved in the process of cell differentiation (Ficz et al., 2011; Ito et al., 2010).
In embryonic stem cells, DNA methylation can also occur at non-CG dinucleotides of genes involved in pluripotency and differentiation. This non-CG methylation is enriched in gene bodies and depleted in protein binding sites and enhancers. However, non-CG methylation is not evident after cellular differentiation occurs (Lister et al., 2009). Because of its role in guiding gene expression patterns during development, 5hmC and non-CG methylation may play a role in the development of psychopathology.
Epigenetic regulation of gene expression can also be accomplished by a variety of noncoding RNA molecules (ncRNAs), which are continuously being identified and characterized (Mattick, Taft, & Faulkner, 2010). These regulatory RNAs can recruit chromatin- or DNA-modifying complexes to restrict the ability of transcription factors and RNA polymerase II to access the transcription start site of a gene. For example, miRNAs are generated by the RNA interference (RNAi) pathway and regulate an extensive array of developmental processes. miRNAs are endogenously expressed RNA hairpins that are cleaved in the nucleus by Drosha, exported to the cytoplasm, and spliced into short segments of 20–25 base pairs by Dicer. This pre-miRNA becomes single stranded when it associates with the RNA-induced silencing complex (RISC). When miRNA binds RISC, it can target the 3′ UTR of mRNA that is complementary to its own sequence (Fig. 10.5). Binding of the miRNA to its complementary mRNA either initiates degradation of the mRNA or directly inhibits translation (Bartel, 2009).
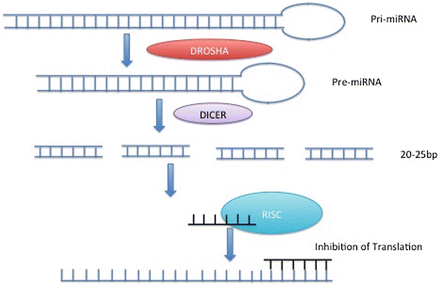
Fig. 10.5
Double-stranded RNA in the nucleus (pri-miRNA) is cleaved by Drosha to produce pre-miRNA. Following export to the cytoplasm, Dicer cleaves the pre-miRNA into 20–25 base pair fragments. When attached to RISC, miRNA can inhibit translation of a gene
Other classes of small RNAs include small interfering RNA (siRNA) that uses miRNA-like mechanism to utilize RISC but silences a gene posttranscriptionally through mRNA cleavage (Taft, Pang, Mercer, Dinger, & Mattick, 2010), piwi-interacting small RNAs (piRNAs) that participate in silencing transposon activity (Ghildiyal & Zamore, 2009), small nucleolar RNAs (snoRNAs) that act like miRNA (Taft, Glazov, Lassmann, et al., 2009), promoter-associated small RNAs (PASRs), and transcription initiation RNAs (tiRNAs) that are linked to transcription start sites and nucleosome positioning (Kapranov et al., 2007; Taft, Glazov, Cloonan, et al., 2009). As such, these regulatory RNAs provide an efficient system for influencing cellular function in a dynamic way.
Epigenetic Mechanisms During Fetal Development
Recent studies have linked preterm delivery and obstetric complications to psychiatric disorders developed later in life. Specifically, those born preterm (<37 weeks of gestation) are more likely to be hospitalized for a psychiatric disorder in adolescence or young adulthood (Lindstrom, Lindblad, & Hjern, 2009; Nosarti et al., 2012), and hypoxia during the prenatal period has been linked to the development of schizophrenia (Nicodemus et al., 2008; Schmidt-Kastner, van Os, Steinbusch, & Schmitz, 2006). Epigenetic regulation participates in several biological phenomena that are essential for an organism’s development (Shi & Wu, 2009) and may underlie abnormal gene expression during neurodevelopment. To explore this hypothesis, we must first explore how epigenetic patterns guide fetal development.
Across the genome, DNA methylation plays a vital role in genomic imprinting, a process in which the expression of a gene occurs in a parent-specific manner. If a gene is maternally imprinted, DNA methylation selectively represses only the copy of the gene contributed by the mother (Abramowitz & Bartolomei, 2012). Thus, only the paternally contributed copy is transcribed. For example, the insulin growth factor 2 (IGF2) gene is maternally imprinted and is only expressed from the paternally contributed copy. IGF2 promotes growth during gestation, and failure of this imprinting mechanism leads to a congenital disorder called Beckwith–Wiedemann syndrome (Chao & D’Amore, 2008).
Following fertilization, areas that are not protected by imprinting undergo both active and passive demethylation (Kafri, Gao, & Razin, 1993; Rougier et al., 1998). DNA methylation patterns then begin to differentiate by developmental stage and by tissue (Liang et al., 2011). DNA methylation also plays a major role in the establishment of gene dosage compensation by inducing heterochromatic repression of gene expression on one of the X chromosomes in female cells (Lucchesi, Kelly, & Panning, 2005). This process is called X chromosome inactivation and results in one of the two X chromosomes in female somatic cells being permanently silenced.
Because fetal development is a period of extensive cellular replication and growth, environmentally induced epigenetic changes may result in stable patterns of gene expression and phenotypic differences among exposed individuals (Dolinoy, Weidman, & Jirtle, 2007; Waterland & Jirtle, 2004; Waterland & Michels, 2007). In contrast, the rate of DNA synthesis is much lower during adulthood, and the epigenome may be more resilient to environmental insults. Thus, early prenatal development is a crucial window for the establishment of long-term epigenetic patterns.
Interruption or alterations of epigenetic programming during developmentally sensitive periods could have negative repercussions on postnatal outcomes that result in life-long consequences. This phenomenon has been demonstrated phenotypically in the work of David Barker, who observed that, during gestations with poor nutrient conditions, low birth weight associates with a higher risk for chronic adult conditions such as stroke, type II diabetes, heart disease, and hypertension (Barker, 1997; Barker & Osmond, 1986). This has come to be known as the developmental origin of health and disease (DOHD) hypothesis and has motivated research linking prenatal factors to differences in postnatal health outcomes. Several independent studies suggest that epigenetic differences can be observed at birth based on both gestational age (Novakovic et al., 2011; Schroeder et al., 2011) and growth patterns (Banister et al., 2011; Tobi et al., 2011), prompting the suggestion that epigenetic changes mediate the relationship between prenatal factors and long-term health problems (Cutfield, Hofman, Mitchell, & Morison, 2007; Menon, Conneely, & Smith, 2012).
Stress Response
The developing brain is particularly sensitive to environmental challenges during periods of developmental plasticity in which personal experiences shape neural circuitry. Several lines of evidence suggest that prenatal or early life stress (ELS) results in epigenetic programming that influences behavior throughout the lifespan. The first postnatal week to 10 days of a newborn rat pup is in many ways developmentally equivalent to the third trimester of a human fetus (Dobbing & Sands, 1979). Therefore, researchers can use rat models to study the effects of perinatal stress on offspring by exposing newborn pups to controlled, stress-inducing environments to gain insight into the potential impact of stress during developmentally sensitive periods.
One such model examined the effects of ELS on methylation by using mothers that exhibited a high level of licking and grooming (LG) and arched back nursing (ABN) behavior compared with those that did not (Weaver et al., 2004). These behaviors are part of a normal maternal nurturing pattern in rodents for mothers nursing neonates. Rats raised by either a natural or surrogate mother with “low” LG/ABN behaviors show stronger HPA axis responses to stress than those raised by “high” LG/ABN mothers (Francis, Diorio, Liu, & Meaney, 1999; Liu et al., 1997).
Rat pups raised by a “low” LG/ABN mother subsequently reveal higher hippocampal methylation of the glucocorticoid receptor (Nr3c1) promoter (NOTE: lower case gene symbol is indicative of an animal study while upper case symbols are used for human studies); methylation of these CpG sites disrupts the binding site of nerve growth factor-inducible protein A (Ngf1-a) and decreases Nr3c1 expression (Weaver et al., 2004). Higher methylation of this promoter also associates with decreased acetylation of histone H3 at Lys 9 (Kadonaga, 1998). Thus, Nr3c1 is a candidate for mediation of long-term health effects due to stress as it encodes the glucocorticoid receptor, a hormone-activated factor that mediates many of the downstream effects of the stress hormone corticosterone. A follow-up study demonstrated that Ngf1-a binding of the Nr3c1 promoter region is responsible for loss of local methylation and may play a role in the epigenetic programming of Nr3c1 (Weaver et al., 2007).
Studies in humans have reported associations between prenatal or ELS and differential methylation patterns of stress-related genes. An increase in umbilical cord blood DNA methylation of the NR3C1 promoter was reported in the offspring of women who experienced a depressive or anxious episode during the second or third trimesters of pregnancy (Oberlander et al., 2008). Increased methylation of a CpG site in the NR3C1 promoter was associated with increased neonatal salivary cortisol response at 3 months of age; this CpG is located in the putative NGFI-A binding site, and these findings resemble results reported in the hippocampus of rats exposed to ELS (Weaver et al., 2004) in that rat pups exposed to lower levels of maternal care exhibited increased DNA methylation of the Nr3c1 promoter and increased corticosterone levels in response to restraint stress.
McGowan and colleagues compared the degree of CpG methylation in the human NR3C1 promoter in DNA extracted from the hippocampus of suicide victims with a history of child abuse, suicide victims without child abuse, and individuals who died in accidents (McGowan et al., 2009). They reported increased methylation of the NR3C1 promoter of the ELS sample. Decreased NGFI-A binding was reportedly correlated with methylation of the NR3C1 promoter suggesting increased methylation at that site decreased NGFI-A-inducible transcription.
Significant stressful events or traumas during early life increase the risk of developing adult psychiatric disorders ranging from posttraumatic stress disorder (PTSD) to substance abuse disorders (Kendler et al., 2000; Mullen, Martin, Anderson, Romans, & Herbison, 1993). Studies in both animals and humans provide considerable strength to the hypothesis that epigenetic factors contribute to cognitive health and psychopathology (Duman & Monteggia, 2006; Roth, Lubin, Sodhi, & Kleinman, 2009; Roth & Sweatt, 2011; Rutter, Moffitt, & Caspi, 2006; Tsankova, Renthal, Kumar, & Nestler, 2007). Along these lines, traumatic events such as child abuse clearly associate with DNA methylation patterns (Beach, Brody, Todorov, Gunter, & Philibert, 2010; McGowan et al., 2009). These findings support the idea that human stress-related “programming” events are associated with DNA methylation and extend the range of these events beyond the in utero period to early life exposure postpartum.
Current Issues
Unlike epigenetic studies of cancer, examination of behavioral and psychiatric traits is complicated by a number of intrinsic factors that limit the interpretations that can be drawn from any single experiment. As such, data derived from multiple complementary approaches, each with their own limitations, are often gathered in support of any particular hypothesis (Ursini et al., 2011).
One of the major challenges in the study of behavioral and psychiatric epigenetics centers on the fact that epigenetic patterns vary by cell type. This issue affects both gene expression and methylation studies, but not studies of sequence variants. While it is important to select a relevant tissue for studying psychiatric disorders, the most relevant tissue is likely to be in the brain, which is not accessible in healthy, living humans. This limitation can be overcome by using animal models, but they often do not adequately reflect the range of symptoms inherent in human psychopathology.
Examination of brain tissue from individuals who have recently died (e.g., postmortem tissue) has its own set of caveats. Often phenotype data associated with these samples must be obtained retrospectively from medical records or by questioning relatives, which may not yield the same quality of information that could be obtained through a prospective design. Because access to these samples is limited, it is difficult to match cases and controls on clinical and demographic factors. Finally, the quality of postmortem tissue is varied based on medical treatment or hypoxia prior to death, postmortem interval, and the storage of the tissue under temperature control in stabilization buffers (Pidsley & Mill, 2011).
Many case–control studies rely on peripheral tissues that are accessible in living humans, such as blood or saliva. These tissues have the added advantage of being amenable to longitudinal studies in which sampling may occur regularly over development of the disorder or its treatment. Peripheral tissues are also most appropriate for biomarker development. Indeed, epigenetic variations in DNA samples from blood have been associated with discrete forms of cancer (Cortese et al., 2012).
The most common peripheral tissue utilized is blood, but the proportion of immune cell types in blood can vary between individuals or by disease state (Bian et al., 2010; Dilli, Oguz, Dilmen, Koker, & Kizilgun, 2010; Gkrania-Klotsas et al., 2010; Hinz et al., 2011; Jafarzadeh, Poorgholami, Izadi, Nemati, & Rezayati, 2010; Singh, 2010). Immune cell types can also vary with exercise level (Santos-Silva et al., 2001) or obesity (Nascimento et al., 2010). Differences in DNA methylation patterns between each of these cell types have been reported for specific genes (Moverare-Skrtic, Mellstrom, Vandenput, Ehrich, & Ohlsson, 2009; Sakamoto, Ortaldo, & Young, 1988) and genome-wide (Sun et al., 2010). Thus, the association between epigenetic variation and any phenotype of interest will likely be confounded by variations in the proportion of cell types within a sample. This caveat applies to brain tissue as readily as to blood as neuronal cells are regulated by a different group of genes than dendritic cells, and efforts should be made to isolate and evaluate a single cell type if possible.
Epigenetic patterns are also influenced by an individual’s ancestry. For example, DNA methylation patterns vary between African American and Caucasian (Adkins, Krushkal, Tylavsky, & Thomas, 2011) though genetic and epigenetic variations are somewhat correlated (Liu et al., 2010). The similarity of methylation patterns in families (Bjornsson et al., 2008) and within twin pairs (Kaminsky et al., 2009) suggests that sequence variation such as single nucleotide polymorphisms (SNPs) or copy number variants (CNVs) may impact methylation patterns. In fact, the phenomenon has been well documented (Kerkel et al., 2008; Schalkwyk et al., 2010; Zhang et al., 2010) though it is not yet clear how this may contribute to any particular disease.
Studies demonstrate that DNA methylation at specific loci can be influenced by sequence variations such that individual genotypes at a given locus may result in different patterns of DNA methylation. These sites have come to be known as sequence-dependent allele-specific methylation (ASM) sites or methylation quantitative trait loci (meQTL). Genotype-methylation correlations can be observed across extended genomic regions (Bell et al., 2010) and have been identified in blood leukocytes, buccal cells, brain tissue, and lymphoblast cell lines (Bell et al., 2010; Li et al., 2010; Schalkwyk et al., 2010; Shoemaker, Deng, Wang, & Zhang, 2010; Zhang, Rohde, Reinhardt, Voelcker-Rehage, & Jeltsch, 2009). Thus sequence variants may be best explored in the context of epigenetic differences (Meaburn, Schalkwyk, & Mill, 2010; Schmidt, Holsboer, & Rein, 2011; Tycko, 2010).
As a specific example, catechol-O-methyltransferase (COMT) encodes an enzyme that plays a key role in the major degradative pathway for several neurotransmitters. This gene contains a common valine (Val) to methionine (Met) substitution at codon 158 (rs4680) that affects the thermostability and activity of COMT and been implicated in mental illness pathophysiology. Met/Met homozygotes exhibit 35–50 % less activity compared to Val/Val homozygotes (Chen et al., 2004), a decrement believed to increase dopamine signaling in Met158 allele carriers (Bilder, Volavka, Lachman, & Grace, 2004; Tunbridge, Bannerman, Sharp, & Harrison, 2004). This substitution also creates a CpG site, and stress-related methylation of this polymorphism has already been linked to brain activity (Ursini et al., 2011).
Because gene expression varies developmentally and across the lifespan, epigenetic studies must consider age as well. Studies have reported widespread age-related methylation differences at CpG sites across the genome (Bjornsson et al., 2008; Bollati et al., 2009; Rakyan et al., 2010; Teschendorff et al., 2010). Some differences appear to be related to the physiological process of aging while others appear to be stochastic. Taken as a whole, the studies described in this section emphasize the importance of using an epidemiological approach in epigenetic studies that carefully matches subjects on demographic and clinical parameters.
Psychopathology
The diagnostic criteria for psychiatric disorders are continually being refined, and individuals with a primary psychiatric diagnosis may still exhibit comorbid traits. Because there are no widely accepted biological tests that delineate one set of behavioral traits from another, many researchers focus on endophenotypes or specific behavioral traits with well-defined etiologies. The following section is not meant to be a comprehensive review of the field to date but will highlight examples and research strategies from ongoing research in mood, anxiety, and psychotic disorders as well as studies of substance abuse.
Mood Disorders
In the United States, as much as 20 % of adults will suffer from a mood disorder during their lifetime, but only a fraction of those will receive adequate treatment (Wittchen, Beesdo, & Bittner, 2003). The most common mood disorders are major depressive disorder (MDD) and bipolar disorder (BPD), and it is widely accepted that both MDD and BPD result from the combined influence of genetic predisposition with adverse experiences, particularly during development.
A study of a genetic variant in the serotonin transporter (SLC6A4) supports this hypothesis (Caspi et al., 2003). The study examined a functional polymorphism in the promoter region of the gene, which is referred to as the serotonin-transporter-linked polymorphic region or the 5-HTTLPR. The short (S) allele is associated with reduced expression of the gene, while the long (L) allele confers normal activity (Lesch et al., 1996). Individuals with the short (S) allele are more likely to have symptoms of depression and suicidality if they also had more stressful life events (Caspi et al., 2003). The results have been controversial because replication of these results is inconsistent (Caspi, Hariri, Holmes, Uher, & Moffitt, 2010; Karg, Burmeister, Shedden, & Sen, 2011; Munafo, Durrant, Lewis, & Flint, 2009; Risch et al., 2009). However, in addition to genotype, the expression of the SLC6A4 gene is also regulated by DNA methylation (Olsson et al., 2010; Philibert et al., 2008; van IJzendoorn, Caspers, Bakermans-Kranenburg, Beach, & Philibert, 2010), further supporting the idea that both sequence variants and epigenetic patterns should be evaluated together in the study of psychopathology.
There are several ways of measuring psychiatric symptoms, and no single method is used consistently across the field. Thus a central research focus is the identification of biological markers (a.k.a. biomarkers) that discriminate between psychiatric diagnosis and symptom state. For depression, one such candidate is brain-derived neurotrophic factor (BDNF), and a recent study suggests that peripheral blood DNA methylation of the BDNF promoter may function as a biomarker for MDD (Fuchikami et al., 2011). Epigenetic regulation of this gene is altered in response to stress (Fuchikami, Yamamoto, Morinobu, Takei, & Yamawaki, 2010), and changes in its expression are implicated in a number of psychiatric disorders (Angelucci, Brene, & Mathe, 2005; Tsankova et al., 2007; Weickert et al., 2003). In this study, individuals with MDD were delineated from those without any history of psychopathology by examining DNA methylation patterns from a blood sample (Fuchikami et al., 2011). Previous studies have noted increased methylation of BDNF in the brain tissue of those who committed suicide (Keller et al., 2010), but peripheral tissues, such as blood, have the greatest utility for clinical testing.
A similar study examined the expression of specific DNMTs, the enzymes that catalyze the addition of a methyl group to DNA, in MDD or BPD individuals (Higuchi et al., 2011). Members of the DNMT family are responsible for establishing and maintaining DNA methylation patterns through cell division (Jeltsch, 2006). Interestingly, the expression levels of some DNMTs were different in those who were in a depressive state when compared to those whose MDD or BPD symptoms were in remission (Higuchi et al., 2011). These studies provide insight into the pathophysiology of mood disorders, and they support the idea that epigenetic factors could be used as biomarkers for diagnosis or symptom severity.
Anxiety Disorders
Fear is a normal emotional response to a perceived threat or danger that is associated with a systemic response throughout the body. For example, anxiety and psychosocial stress correlate with changes in the digestive, cardiovascular, and immune systems (Fetissov & Dechelotte, 2011; Gill, Saligan, Woods, & Page, 2009; Steiner, 2011; Weiss et al., 2011). Anxiety disorders result from an uncommon response to apprehension or anxiety that compromises daily activities. The source of the fear can be generalized (generalized anxiety disorder), specific to an object or situation (phobia), or a result from a past posttraumatic experience (PTSD). PTSD is a debilitating, stress-related psychiatric disorder (Kessler, 2000; Kessler et al., 1994), but because of the specificity of the index event, PTSD is particularly amenable for genetic and epigenetic investigations.
The response to stress is mediated by neural circuits throughout the brain, which activate neuroendocrine and autonomic stress systems (Charmandari, Tsigos, & Chrousos, 2005; Pacak & Palkovits, 2001). However, men and women experience different types of stressors, and common stressors may be processed in different ways. For example, association of the PACAP–PAC1 receptor pathway has been reported in women but not men with PTSD (Ressler et al., 2011). The neuropeptide pituitary adenylate cyclase-activating polypeptide (PACAP) functions in parts of the brain that mediate anxiety and fear-related behaviors. A genetic variant in the PAC1 receptor (ADCYAP1R1) that disrupts a putative estrogen response element associates with PTSD selectively in women. Further, PTSD associates with peripheral blood levels of PACAP in females and also with alterations in peripheral blood DNA methylation and mRNA expression of the ADCYAP1R1 transcript. These data suggest that ADCYAP1R1 is regulated, in part, through epigenetic mechanisms that contribute to differential function of the PAC1 receptor in women with PTSD.
Studies have also linked epigenetic patterns in individuals with PTSD to dysregulation of the immune system. Multiple studies have identified DNA methylation differences in genes related to immune function and inflammation of those with PTSD compared to controls (Smith et al., 2011; Uddin et al., 2010). Consistent with this, individuals with PTSD have increased levels of circulating inflammatory cytokines (Maes et al., 1999; Spivak et al., 1997; von Kanel et al., 2007) and lower anti-inflammatory cytokines (Kawamura, Kim, & Asukai, 2001; von Kanel et al., 2007). Together, these results suggest that psychosocial stress may alter global and gene-specific DNA methylation patterns potentially associated with peripheral immune dysregulation. Studies in this area not only contribute to our understanding of the communication between the nervous and immune systems but also further support the possibility of peripheral testing for psychiatric disorders.
Psychotic Disorders
Schizophrenia is characterized by the presence of psychotic symptoms, which include disorganized thought, paranoia, hallucinations, and delusions. These symptoms are accompanied by affective flattening responses and by altered cognitive abilities. They are also accompanied by epigenetic changes in the brain. For example, the total amount of miRNAs and expression of specific miRNAs increase in the cerebral cortex of postmortem samples from schizophrenia patients compared to controls (Beveridge, Gardiner, Carroll, Tooney, & Cairns, 2010; Gavin & Akbarian, 2012). While miRNAs regulate a wide variety of genes, miRNAs implicated in schizophrenia also regulate genes that participate in neurodevelopment, which may have particular relevance for the hypothesis that prenatal events confer susceptibility to schizophrenia (Wong et al., 2013). Examination of differentially expressed miRNA target genes has also provided insight into the results of both focused and large-scale genetic studies (Kim et al., 2012; Kwon, Wang, & Tsai, 2013).
In a separate postmortem examination of individuals with schizophrenia, DNA methylation in the frontal cortex occurs in genes that are linked to disease etiology including glutamatergic and GABAergic neurotransmission, stress response, and brain development (Mill et al., 2008). Interestingly, DNA methylation changes in this study were also enriched in regions of the genome identified in linkage studies of schizophrenia. Like previous examinations of PTSD, Mill and colleagues not only identified epigenetic differences common to both sexes, but they also identified differences specific to males and females.
Postmortem studies are often confounded by differences in age, race, or other environmental exposures. To control for such factors, investigations of monozygotic (a.k.a. identical) twins that are discordant for schizophrenia can be used as an alternative to unrelated individuals. While these studies tend to have smaller number of subjects, the genetic and environmental variation between each co-twin is limited and results in a powerful design to study epigenetic effects. One recent study reported considerable epigenetic variation between twins discordant for schizophrenia. Interestingly, they then compared these differences to twins discordant for BPD, who also experience psychotic symptoms. While some epigenetic differences were specific to either schizophrenia or BPD, some were consistently altered in both disorders (Dempster et al., 2011).
Substance Abuse
Substance abuse is a chronic and relapsing brain disease that causes overdependence on a particular substance, such as alcohol or cocaine, despite negative consequences. These substances artificially activate the brain’s reward circuitry and result in mood changes and health problems. Because the cause and effect of this disorder are reasonably well understood, it can be effectively modeled in animals to isolate the effects of independent substances and explore the neurobiology of tolerance, dependence, withdrawal, craving, and relapse.
Alcohol abuse affects multiple tissues and causes widespread epigenetic changes (Shukla et al., 2008). For example, alcohol causes acetylation of histone H3 at Lys 9 (H3K9) in hepatocytes (Park, Miller, & Shukla, 2003). This increase in H3K9 acetylation correlates with increased expression of alcohol dehydrogenase (ADH1), a subunit of the enzyme that metabolizes ethanol (Park, Lim, & Shukla, 2005). Alcohol-dependent changes in the brain are also evident. Primary cortical neurons exposed to chronic intermittent ethanol have extensive changes in miRNA expression, which regulate gene expression through posttranscriptional mechanisms (Guo, Chen, Carreon, & Qiang, 2012). Some of these miRNAs target CpG islands or specific genes, and, as a group, they are enriched for the pathways that regulate transcription, neuron differentiation, embryonic development, protein phosphorylation, and regulation of synaptic plasticity.
Psychotropic Medications
It is difficult to interpret whether observed epigenetic differences represent a causal factor in behavior or psychopathology. Epigenetic correlates in clinical populations may be attributable to environmental factors that increase risk for developing psychopathology or potentially to the symptoms or treatment of the disorder. However, increasing evidence demonstrates that commonly used psychotropic medications alter epigenetic patterns independently. Few studies are capable of discerning whether epigenetic changes associate with medication exposure or the disorder itself. Ideally, a well-controlled experiment should gather data from groups of ill subjects who are untreated as well as treated with only the medication of interest and compared to groups of treated and untreated subjects who do not have the disorder. However, it is not ethical to withhold treatment from those who are ill, and side effects may preclude medication use in those who are not ill.
Extended exposure to antipsychotic medications may result in specific DNA methylation differences not associated with the disorder that they are being used to treat (Mill et al., 2008). In the case of schizophrenia and BPD, disorders for which antipsychotics are often prescribed, animal studies can provide insight. For example, evaluation of clozapine and haloperidol, two atypical or second-generation antipsychotics, revealed that treatment with clozapine, but not haloperidol, increases histone 3-lysine 4 (H3K4) trimethylation and H3 acetylation while decreasing DNA methylation of the promoters of treatment-related genes, such as reelin and glutamic acid decarboxylase (Gad67) in the frontal cortex (Dong, Nelson, Grayson, Costa, & Guidotti, 2008; Huang et al., 2007). While the exact mechanism through which clozapine promotes chromatin remodeling and changes in synaptic plasticity is unknown, chromatin remodeling may begin with histone acetylation and subsequent recruitment of proteins involved in DNA demethylation.
Related posts:
 Sleep and Psychopathology: Quantitative and Molecular Genetic Research on Comorbidity
Genetic Influences on Depression and Anxiety in Childhood and Adolescence
Progress in Understanding the Causes of Autism Spectrum Disorders and Autistic Traits: Twin Studies from 1977 to the Present Day
Sleep and Psychopathology: Quantitative and Molecular Genetic Research on Comorbidity
Genetic Influences on Depression and Anxiety in Childhood and Adolescence
Progress in Understanding the Causes of Autism Spectrum Disorders and Autistic Traits: Twin Studies from 1977 to the Present Day
 Genetics of Substance Use Disorders
Genetics of Substance Use Disorders
 Additional Evidence for Meaningful Etiological Distinctions Within the Broader Construct of Antisocial Behavior
Additional Evidence for Meaningful Etiological Distinctions Within the Broader Construct of Antisocial Behavior
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


