Such trans-generational epigenetic transmission is likely to vary with the number of subsequent generations. For example, certain epigenetic modifications of histones in the chromatin of Caenorhabditis elegans parents, known to affect their longevity, are not entirely erased during gametogenesis and, therefore, can also extend the lifespan of the offspring up to the third generation (Greer et al. 2011). In this case, this trans-generational inheritance appears to affect preferentially the epigenetic regulation (expression) of genes involved in metabolic pathways (Greer et al. 2011).
What are the sources of inter-individual variability in the epigenome? In the next two sections, we will discuss stochastic instability in the transfer of epigenetic markers during mitosis and environment-induced epigenetic modifications (see Fig. 5.2).
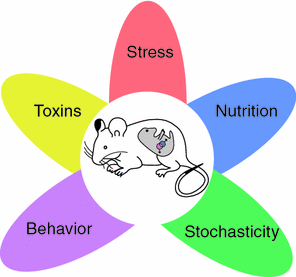
Fig. 5.2
Sources of epigenetic variability. Five environmental influences that affect the developing embryo and its primordial germ cells (represented by the pink and blue dots). Adapted based on Faulk and Dolinoy (2011)
5.1.2 Stochastic Instability
Epigenetic marks are transmitted from cell to daughter cell during mitosis and, to a much lesser extent, from a parent to offspring through the germ line. This transfer of epigenetic information involves a variety of molecular processes (see Sect. 5.2 for details) and, not surprisingly, it is prone to errors. Thus, it has been estimated that the error rate for replicating epigenetic marks is ~1 in 1,000; this is much higher than the estimated error rate during the DNA replication [~1 in 1,000,000 bases; Hjelmeland (2011)]. These errors introduce a certain level of randomness into the transmission of epigenetic information. Stochastic instability then refers to the probabilistic nature of the processes that results both from a predictable action and the random element. The above example of agouti mice illustrates the range of such stochastic instability: the level of DNA methylation of the “metastable epiallele” can vary by over 80 % across the isogenic mice; a phenomenon stable during the life of an individual but stochastic across different individuals (Dolinoy et al. 2010). What are the main sources of the predictable actions and the most important time window of their influence on the epigenome?
5.1.3 Environment-Induced Epigenetic Modifications
During gametogenesis, epigenetic marks are first (largely) erased and then re-established de novo. Thus, in utero, the re-establishment of epigenetic marks can be influenced by a variety of environmental influences acting on the pregnant mother (F0 generation) and both the somatic and germ lines of the embryo (F1 generation). In the case of the former, such somatic “epimutations” will be transmitted from cell to daughter cell throughout the prenatal and post-natal life of the exposed (F1) offspring; these effects are likely to be present in all tissues. In the case of the latter, the germ-line epimutations have the potential for being transmitted to the subsequent (F2) generation, and beyond (see Fig. 1.3). On the other hand, if the environment acts post-natally, its effect is more likely to be seen in specific tissues; thus, tobacco smoke is more likely to affect lung tissue, and diet (together with local bacteria; see the paragraph on “body environment” in Sect. 3.1) is more likely to affect the gut tissue. Figure 5.2 illustrates the most common exposures that have been investigated in the context of environment-induced changes in epigenome; these vary from the effects of specific diets, toxins, stress and behaviour (reviewed in Faulk and Dolinoy 2011). One of the most exiting models of behaviour-induced epigenetic modifications has been introduced by Michael Meaney and his colleague; in a series of experiments, they showed that high (vs. low) levels of licking and grooming (by the dam) during the first week of pups’ lives is associated with lower levels of methylation of the promoter of the glucocorticoid receptor (and, in turn, its higher expression) in the hippocampus, as well as lower response of the hypothalamic-pituitary-adrenal axis to stress (reviewed in Zhang et al. 2013; see Fig. 5.3).
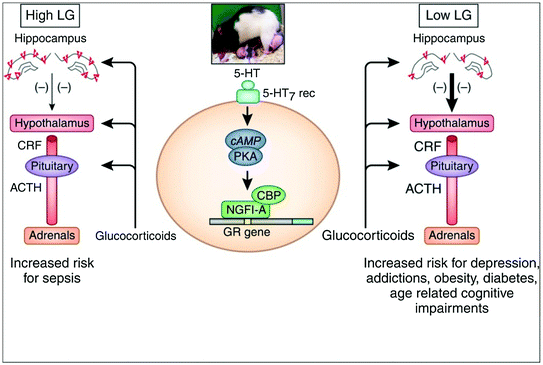
Fig. 5.3
Associations between maternal behaviour (licking and grooming), expression of the glucocorticoid receptor in the hippocampus, regulation of the hypothalamus-pituitary-adrenal axis and psychopathology (right side). LG licking and grooming, ACTH adrenocorticotropin, CRF corticotropin releasing factor, 5-HT serotonin, cAMP cyclic adenosine monophosphate, PKA protein kinase A, NGFI-A nerve growth factor–inducible factor A, CBP CREB-binding protein, GR glucocorticoid receptor. From Zhang et al. (2013)
As pointed out above, epigenetic modifications involve a variety of mechanisms, including DNA methylation, modifications of histone proteins and post-transcriptional modifications of non-coding RNA, such as microRNAs, which regulate translation of mRNA into polypeptides. I will now describe two of these mechanisms in some detail.
5.2 DNA Methylation and Histone Modifications
The methylation of cytosine is the most common epigenetic modification of DNA. Given that the diploid human genome contains 6 × 109 nucleotides (A, C, G and T), there are about 150,000,000 cytosines that—in theory—can exist, in either a methylated or unmethylated state. But, in fact, methylation takes place most often (but not exclusively) when cytosine sits next to guanine—that is, if it is present as CpG4 dinucleotides; when CpG dinucleotides cluster together, we talk about CpG islands. Note that about 60 % of human gene promoters are associated with CpG islands, thus providing a powerful means for DNA methylation to influence gene expression (Portela and Esteller 2010). The so-called CpG shores (areas flanking the CpG islands) show higher variation in DNA methylation, even though the CpG density is lower in these regions as compared with the CpG islands (Rakyan et al. 2011). As shown in Fig. 5.4, DNA methylation can occur not only in CpG islands and shores but also in the gene body, and on repetitive sequences, including those associated with transposable elements (see above for the agouti mice). In general, methylation of the CpG islands at promoters of genes is associated with a reduced rate of DNA transcription. On the other hand, methylation in the gene body facilitates gene expression by preventing spurious initiations of transcription (Portela and Esteller 2010).
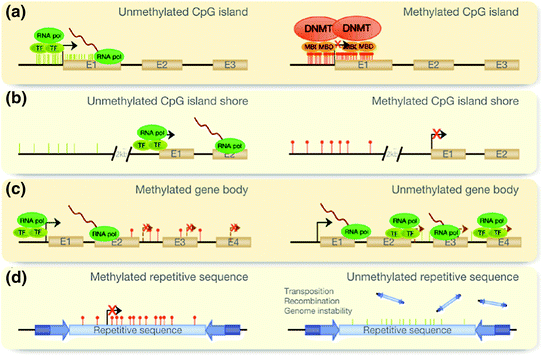
Fig. 5.4
DNA methylation. “DNA methylation can occur in different regions of the genome. The alteration of these patterns leads to disease in the cells. The normal scenario is depicted in the left column and alterations of this pattern are shown on the right. a CpG islands at promoters of genes are normally unmethylated, allowing transcription. Aberrant hypermethylation leads to transcriptional inactivation. b The same pattern is observed when studying island shores, which are located up to 2 kb upstream of the CpG island. c However, when methylation occurs at the gene body, it facilitates transcription, preventing spurious transcription initiations. In disease, the gene body tends to demethylate, allowing transcription to be initiated at several incorrect sites. d Finally, repetitive sequences appear to be hypermethylated, preventing chromosomal instability, translocations and gene disruption through the reactivation of endoparasitic sequences. This pattern is also altered in disease.” E Exon, TF transcription factor, RNA pol RNA polymerase, DNMT DNA methyltransferase, MBD methyl-CpG-binding-domain proteins. From Portela and Esteller (2010)
How does DNA methylation regulate gene expression? One of the mechanisms involves recruitment of special proteins5 that go on to recruit histone-modifying and chromatin-remodelling complexes to the methylated site. As explained below, the DNA-chromatin complex is a powerful regulator of gene expression by virtue of “opening” and “closing” DNA for transcription (see Sect. 4.1). Another mechanism involves direct inhibition of transcription by preventing the binding of relevant transcription factors to the promoter (e.g. Kuroda et al. 2009).
Recall that DNA is packaged with protein complexes to form chromatin. Heterochromatin contains tightly packed and inactive DNA, whereas euchromatin contains a stretched out and active DNA molecule ready for transcription (Sect. 4.1). Histone proteins are the main actors in the transformation of heterochromatin to euchromatin and back (see Fig. 4.1). Gene expression requires uncoiling of chromatin fibres, a process guided by H1 histone and its bonds with DNA molecules. Once uncoiled, two turns of DNA molecule are wrapped around an octamer of core histones (H2, H3A, H3B and H4) in the individual nucleosomes; the N-terminal tails of the core histones protrude out of a nucleosome (see Fig. 4.1b). Importantly, various chemical processes6 can modify amino acids in these histone tails leading, in turn, to the binding of different proteins, affecting local condensation of chromatin and, as such, the level of transcriptional activity at this particular stretch of DNA molecule.7
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


