Chapter 11 Epilepsies Due to Monogenic Disorders of Metabolism
This chapter describes a number of inherited metabolic disorders in which epilepsy is a prominent symptom (Table 11-1). These specific disorders were selected among the large group of metabolic diseases for one or more of the following reasons: (1) relatively high prevalence, (2) presentation in adulthood, (3) knowledge of the underlying genetic defect, and (4) availability of a treatment. Disorders that are very rare, that present exclusively in infancy or childhood, in which epilepsy is uncommon, and for which there is no known genetic defect and no specific treatment will not be discussed.
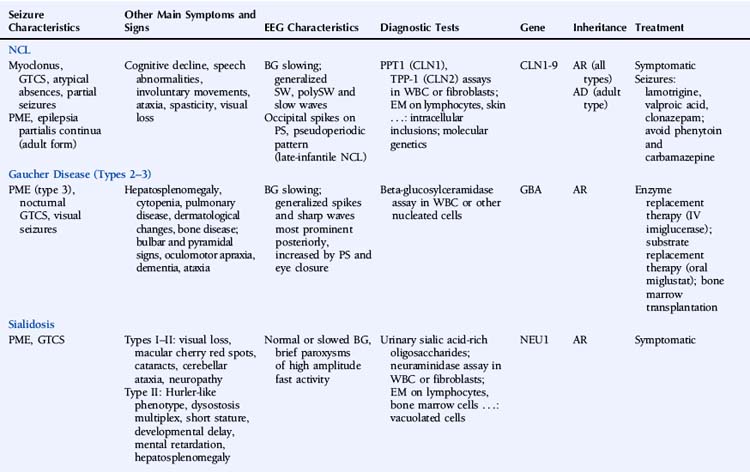

Neuronal Ceroid Lipofuscinoses
Epilepsy is a feature of all types of NCL and is usually the first symptom of disease in the late-infantile form.1 Seizures can manifest as myoclonic jerks, generalized tonic-clonic seizures (primary or secondarily), atypical absences, or partial seizures. Adult NCL may present as a progressive myoclonic epilepsy2 or as epilepsia partialis continua.3 Northern epilepsy is characterized by childhood-onset generalized tonic-clonic or complex partial seizures decreasing after puberty and not accompanied by myoclonus.4
The EEG is characterized by slowing of background activity and the presence of generalized spike-wave and polyspike-wave complexes and bursts of slow waves.5 High-voltage, polyphasic spikes in the occipital region with photic stimulation at 1 to 2 Hz, as well as a pseudoperiodic pattern, have been described in the late-infantile form.6,7
Brain magnetic resonance imaging (MRI) demonstrates cerebral and cerebellar atrophy, T2-hyperintensity of the lobar white matter, thinning of the cerebral cortex, and thalamic T2-hypointensity.8
The diagnostic testing strategy in the NCLs depends mainly on the age of onset. Diagnosis is based on enzymatic assays, electron microscopy, and molecular genetic testing. Two types of enzymatic deficiency have been identified in the NCLs. Palmitoyl-protein thioesterase 1 (PPT1) activity is absent in NCLs caused by mutations in the CLN1/PPT1 gene, and tripeptidyl-peptidase 1 (TPP-1) activity is usually absent in NCLs caused by mutations of the CLN2/TPP1 gene. Enzymatic assays can be performed on leukocytes or fibroblasts. Enzymatic deficiencies in the other NCLs are unknown. Electron microscopy of lymphocytes and tissue biopsies (usually skin) shows typical intracellular inclusions consisting of autofluorescent lipopigment storage material. Eight genes—CLN1/PPT1, CLN2/TPP1, CLN3, CLN5, CLN6, CLN7/MFSD8, CLN8, and CLN10/CTSD—are known to be associated with NCL.9 Numerous different mutations have been identified, but most genes carry a small number of common mutations. The genes at the CLN4 ad CLN9 loci have not been identified. The NCLs are characterized by extensive phenotypic and genetic heterogeneity.
Therapy for the NCLs is limited to symptomatic treatment at present. Promising future treatments include enzyme replacement therapy, gene therapy, and stem cell therapy.10 Lamotrigine has been reported to be effective and well tolerated in the infantile and juvenile forms.11–12 In juvenile NCL, valproic acid is a valuable alternative, and clonazepam may be useful as add-on therapy.13 Carbamazepine and phenytoin should be avoided, as they may increase seizure activity.14,15
Gaucher Disease
Gaucher disease (GD) is an autosomal recessive lysosomal disorder caused by a deficiency of the enzyme beta-glucocerebrosidase (also called acid beta-glucosidase or acid beta-glucosylceramidase) and accumulation of glucosylceramide (GL1) and other glycolipids. Three major clinical subtypes (1, 2, and 3) and two other subtypes (perinatal lethal and cardiovascular) are recognized based on clinical presentation. Type 1 (nonneuronopathic form) usually has no primary central nervous system symptoms. Types 2 (acute neuronopathic form) and 3 (subacute neuronopathic form) are characterized by the presence of primary neurologic disease. They are classically distinguished by age of onset and rate of disease progression, but it is now increasingly recognized that neuronopathic GD represents a phenotypic continuum, ranging from severely affected infants to asymptomatic adults. GD prevalence estimates vary between 1/57,000 and 1/86,000.16,17
GD is a multisystemic disorder characterized by varying degrees of hematological, skeletal, pulmonary, and neurological involvement. Hepatosplenomegaly usually precedes neurological manifestations. Neurological symptoms may include bulbar and pyramidal signs, oculomotor apraxia, dementia, and ataxia. Epilepsy is mainly a symptom of type 3 GD and usually presents as progressive myoclonic epilepsy (also called type 3a GD).18 The myoclonus may be spontaneous, stimulus sensitive, or induced by action. Other reported seizure types include generalized tonic-clonic seizures during sleep and visual seizures.19,20
EEG shows gradual background slowing and generalized spikes and sharp waves, most prominent over the posterior regions and increased by photic stimulation and eye closure.20,21 Brain MRI may show mild cerebral atrophy.
The GBA gene is the only gene known to be associated with GD. At least 200 GBA mutations have been identified.22,23 Four common mutations account for the majority of cases. Molecular genetic testing in a proband is not necessary to confirm the diagnosis, but may be considered for genetic counseling purposes, primarily for carrier detection among at-risk relatives. Genotype-phenotype correlation in GD is poor. Although the genotypic spectrum in patients presenting with progressive myoclonic epilepsy is different from that in other patients with type 3 GD, there appears to be no specific shared genotype.24
Affected individuals should be monitored regularly, including medical history, physical and neurological examination, blood tests—especially hemoglobin concentration and platelet count—assessment of spleen and liver volumes, screening for pulmonary hypertension, and skeletal involvement. Enzyme replacement therapy with imiglucerase, an intravenous recombinant glucosylceramidase enzyme preparation, can reverse systemic involvement. The effectiveness of enzyme replacement therapy for the treatment of neurologic disease remains to be established, although a few reports have suggested some benefit.25–27 Onset of progressive myoclonic seizures while on enzyme replacement therapy appears to indicate a poor prognosis.28 Substrate reduction therapy with the oral agent miglustat is another treatment option in individuals with mild to moderate GD for whom enzyme replacement therapy is not a therapeutic option. A case report described neurologic improvement in a patient with type 3 GD and myoclonic epilepsy on combined enzyme replacement and substrate reduction therapy.29 Individuals with chronic neurologic GD and progressive disease despite enzyme replacement therapy may be candidates for bone marrow transplantation.
Sialidosis
Type I sialidosis (also known as the “normosomatic” type or cherry red spot–myoclonus syndrome) usually presents in the second or third decade and is characterized by progressive visual loss, bilateral macular cherry red spots, cataracts, progressive generalized myoclonus, generalized tonic-clonic seizures, and cerebellar ataxia. The myoclonus is often induced by action. Peripheral neuropathy has also been reported.30 Intellect is usually preserved. Type II sialidosis (also known as the “dysmorphic” type) is the more severe, early onset form and is additionally associated with a Hurler-like phenotype, dysostosis multiplex, short stature, developmental delay, mental retardation, and hepatosplenomegaly.
Reports of EEG observations in sialidosis are rare. Brief paroxysms of high amplitude fast activity on a normal or slightly slowed background were described in one study.31 Jerk-locked back-averaging shows a consistent temporal relationship between the EEG spikes and myoclonic jerks.32 Brain MRI shows progressive cerebral and pontocerebellar atrophy.33
The sialidoses are caused by mutations in the NEU1 gene. Its product, neuraminidase or lysosomal sialidase, has a dual physiologic function: it participates in intralysosomal catabolism of sialated glycoconjugates and is involved in cellular immune response. More than 40 different mutations have been characterized.34–36 In general, there is a close correlation between the residual enzyme activity and the clinical disease severity.37
Treatment is symptomatic. At present, there is no disease-specific treatment available, but potential treatment strategies such as gene therapy and enzyme replacement therapy are under study.38 Treatment of seizures and myoclonus is aspecific. One report described successful treatment of myoclonus with 5-hydroxytryptophan as add-on therapy.39
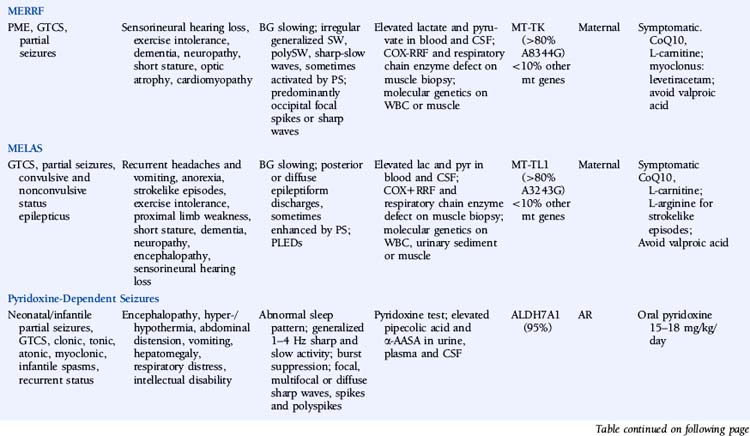
Myoclonic Epilepsy with Ragged-Red Fibers (MERRF)
The disease is characterized by myoclonus, which is often the first symptom, followed by epilepsy and ataxia. Seizures are usually generalized myoclonic or tonic-clonic, but partial seizures have been reported in atypical cases.40 Other common manifestations include sensorineural hearing loss, exercise intolerance, dementia, peripheral neuropathy, short stature, optic atrophy, and cardiomyopathy. Pigmentary retinopathy, ophthalmoparesis, pyramidal signs, and multiple lipomas are occasionally observed. The disease manifestations are very heterogeneous, and overlap syndromes between MERRF and mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes (MELAS) have been reported.41
Lactate and pyruvate levels in blood and cerebrospinal fluid (CSF) are commonly elevated at rest and increase excessively after moderate activity. EEG findings include slowing of background activity and bursts of atypical, irregular generalized spike-wave complexes, polyspike-wave complexes or sharp-slow wave complexes, sometimes activated by photic stimulation.40,42,43 These discharges are often, but not always, related to generalized myoclonic jerks. Focal spikes or sharp waves are also seen, most commonly over the occipital regions. Brain MRI often shows brain atrophy and basal ganglia calcification. Muscle biopsy shows typical cytochrome oxidase (COX) negative ragged-red fibers. Biochemical studies in muscle usually show defects in respiratory chain enzyme activity, especially COX deficiency, but may occasionally be normal. The diagnosis is confirmed by molecular genetic testing, demonstrating mutations in the mitochondrial DNA (mtDNA) gene MT-TK, encoding tRNALys. Over 80% of affected individuals with typical findings carry the A8344G mutation.44 Three additional mutations (T8356C, G8363A, and G8361A) account for about 10% of affected individuals. The mutations appear to exert their pathogenic effects through impairment of protein synthesis.45 The remaining 10% of affected individuals may have other mutations in MT-TK or mutations in a number of other mitochondrial genes. Mutations are usually present in all tissues and can thus be detected in mtDNA from blood leukocytes. However, the occurrence of heteroplasmy (differences in cellular mutational load) can result in variations of tissue distribution of mutated mtDNA. Hence, in some cases the mutation may be undetectable in leukocytes and may only be detected in other tissues, most reliably in skeletal muscle. For the same reason, accurate prediction of phenotype in oligosymptomatic individuals or at-risk family members based on test results is not possible.
There is no specific treatment for the disease. Empirical treatment with coenzyme Q10 (50 to 100 mg 3x/day) and L-carnitine (1000 mg 3×/day), aiming to improve mitochondrial function, is often used.46 No controlled studies have compared the efficacy of various AEDs in MERRF. Valproic acid should be used with caution because of the increased risk of hepatotoxicity.47 A number of reports have described substantial improvement of myoclonus with levetiracetam.48,49 Potential future treatments for MERRF include selective inhibition of mutant mtDNA replication by peptide nucleic acids and import of nuclear-encoded tRNALys into mitochondria.50,51
Mitochondrial Encephalomyopathy, Lactic Acidosis, and Strokelike Episodes (MELAS)
MELAS is another disorder caused by mutations in mtDNA and thus is transmitted by maternal inheritance. Onset is typically in childhood, though infantile and adult onset has been reported. The most common presenting symptoms are seizures, recurrent headaches, anorexia, and recurrent vomiting. Seizures can be generalized or partial and are often associated with strokelike episodes of transient hemiparesis or cortical blindness. Generalized convulsive and complex partial status epilepticus, as well as epilepsia partialis continua, have been reported.52–54 Other common symptoms and signs include exercise intolerance, proximal limb weakness, short stature, encephalopathy, dementia, sensorineural hearing loss, and peripheral neuropathy. Less-common symptoms include myoclonus, ataxia, episodic coma, optic atrophy, cardiomyopathy, pigmentary retinopathy, ophthalmoplegia, diabetes mellitus, hirsutism, gastrointestinal dysmotility, and nephropathy. MELAS should be suspected based on the following features: (1) strokelike episodes before age 40; (2) encephalopathy with seizures and/or dementia; and (3) lactic acidosis and/or ragged-red fibers on muscle biopsy, or both. The diagnosis may be confirmed if at least two of the following are also present: normal early psychomotor development, recurrent headache, or recurrent vomiting.55 There is a wide variability in clinical presentation, and overlap syndromes between MELAS and other mitochondrial disorders have been reported.
Lactate and pyruvate levels in blood and CSF are commonly elevated at rest and increase excessively after moderate activity. EEG commonly shows slowing of alpha-rhythm and posterior or diffuse epileptiform discharges, sometimes enhanced by photic stimulation.40,56–58 Periodic lateralized epileptiform discharges (PLEDs) with alternating focus have been reported during episodes of complex partial status epilepticus.52,59 Basal ganglia calcifications are commonly seen on computed tomography (CT). Brain MRI during strokelike episodes shows cortico-subcortical T2- and FLAIR-hyperintense lesions in the posterior regions that slowly spread in the weeks following initial symptoms.60–62 Diffusion-weighted MRI (DWI) demonstrates increased apparent diffusion coefficient (ADC) values in these lesions, distinguishing them from classic ischemic strokes.61 Muscle biopsy typically shows COX-positive ragged red fibers and an abundance of mitochondria. Biochemical analysis of respiratory chain enzymes in muscle usually shows multiple partial defects, but it can also be normal. The diagnosis is confirmed by molecular genetic testing, demonstrating mutations in the mtDNA gene MT-TL1, encoding tRNALeu. tRNALeu is essential for mitochondrial protein synthesis, specifically for the incorporation of leucine into nascent proteins. The A3243G mutation is present in over 80% of individuals with typical clinical findings.63 The remainder of cases are caused by other mutations in MT-TL1 or mutations in other mtDNA genes, most commonly MT-ND5. Mutations are usually present in all tissues and can thus be detected in leukocytes. However, the percentage of mutated mtDNA decreases in blood with age in patients harboring the A3243G mutation,64 and in some cases the pathogenic mutation may be undetectable in leukocytes because of heteroplasmy. In such cases, urinary sediment has proven the most useful among accessible tissues for detecting the A3243G mutation.65,66 A muscle biopsy is recommended in the rare instance in which the MT-TL1 A3243G mutation cannot be detected by standard techniques in leukocytes or urinary sediment from an individual with classic MELAS. Accurate prediction of phenotype in oligosymptomatic individuals or at-risk family members based on genetic test results is not possible.
No specific treatment for MELAS exists. Coenzyme Q10 (50 to 100 mg 3×/day) and L-carnitine (1000 mg 3×/day) may be of some benefit. L-arginine has been reported to improve strokelike symptoms when given in the acute phase and to decrease frequency and severity of strokelike episodes when given between episodes.67,68 Beneficial effects of oral succinate were reported in one individual.69 Seizures usually respond to traditional AEDs, though valproic acid may aggravate seizures.70,71
Related posts:
 The Life-Threatening Epilepsies of Childhood and Their Treatment
The Life-Threatening Epilepsies of Childhood and Their Treatment
 The Spectrum of Epilepsies Associated with Generalized Spike-and-Wave Patterns
The Spectrum of Epilepsies Associated with Generalized Spike-and-Wave Patterns
 The Surgery of Temporal Lobe Epilepsy I—Historical Development, Patient Selection, and Seizure Outcome
The Surgery of Temporal Lobe Epilepsy I—Historical Development, Patient Selection, and Seizure Outcome
 Brain Stimulation in Epilepsy—An Old Technique with a New Promise?
Brain Stimulation in Epilepsy—An Old Technique with a New Promise?
 Cortical Myoclonus and Epilepsy: Overlap and Differences
Cortical Myoclonus and Epilepsy: Overlap and Differences
 Does Early Treatment Influence the Long-Term Outcome of Epilepsy?
Does Early Treatment Influence the Long-Term Outcome of Epilepsy?



