Electro-clinical syndromes arranged by age at onset a
Neonatal period
Benign familial neonatal epilepsy (BFNE)
Early myoclonic encephalopathy (EME)
Ohtahara syndrome
Infancy
Epilepsy of infancy with migrating focal seizures
West syndrome
Myoclonic epilepsy in infancy (MEI)
Benign infantile epilepsy
Benign familial infantile epilepsy
Dravet syndrome
Myoclonic encephalopathy in non-progressive disorders
Childhood
Febrile seizures plus (FS+) (can start in infancy)
Panayiotopoulos syndrome
Epilepsy with myoclonic atonic (previously astatic) seizures
Benign epilepsy with centrotemporal spikes (BECTS)
Autosomal-dominant nocturnal frontal lobe epilepsy (ADNFLE)
Late onset childhood occipital epilepsy (Gastaut type)
Epilepsy with myoclonic absences
Lennox-Gastaut syndrome
Epileptic encephalopathy with continuous spike-and-wave during sleep (CSWS)b
Landau–Kleffner syndrome (LKS)
Childhood absence epilepsy (CAE)
Adolescence – Adult
Juvenile absence epilepsy (JAE)
Juvenile myoclonic epilepsy (JME)
Epilepsy with generalized tonic–clonic seizures alone
Progressive myoclonus epilepsies (PME)
Autosomal dominant epilepsy with auditory features (ADEAF)
Other familial temporal lobe epilepsies
Less specific age relationship
Familial focal epilepsy with variable foci (childhood to adult)
Reflex epilepsies
Distinctive constellations
Mesial temporal lobe epilepsy with hippocampal sclerosis (MTLE with HS)
Rasmussen syndrome
Gelastic seizures with hypothalamic hamartoma
Hemiconvulsion–hemiplegia–epilepsy
Epilepsies that do not fit into any of these diagnostic categories can be distinguished first on the basis of the presence or absence of a known structural or metabolic condition (presumed cause) and then on the basis of the primary mode of seizure onset (generalized vs. focal)
Epilepsies attributed to and organized by structural-metabolic causes
Malformations of cortical development (hemimegalencephaly, heterotopias, etc.)
Neurocutaneous syndromes (tuberous sclerosis complex, Sturge–Weber, etc.)
Tumor
Infection
Trauma
Angioma
Perinatal insults
Stroke
Etc.
Epilepsies of unknown cause
Conditions with epileptic seizures that are traditionally not diagnosed as a form of epilepsy per se
Benign neonatal seizures (BNS)
Febrile seizures (FS)
In addition, the ILAE classification recognizes clinical entities not strictly considered syndromes but representing clinically distinctive constellations due to specific lesions or other causes. Examples include mesial temporal lobe epilepsy associated with hippocampal sclerosis, gelastic seizures of hypothalamic hamartoma, and Rasmussen epilepsy.
Further, structural/metabolic epilepsies are determined by specific lesions, such as malformations of cortical development, neurocutaneous syndromes, tumors, vascular abnormalities, trauma, and so on.
Finally, epilepsies of unknown causes are recognized, encompassing all epilepsies that in the past were termed “cryptogenic” (i.e., the majority of epilepsy with focal seizures of adulthood).
24.3 Epidemiology
The annual incidence of newly diagnosed epilepsy is 44 per 100,000 person-year in the United States (Rochester, Minnesota) [3] and between 33 and 68 in Europe [4]. The prevalence of active epilepsy is between 4 and 10 per 1000, with a slight, rarely significant predominance in males [4]. Most studies show a predominance of focal (60–70 %) over generalized seizures. The risk for epilepsy is greater in the first years of life, declines thereafter, and increases again in later years: the highest prevalence for epilepsy (9.4 per 1000) is in people aged 85 years or more. The lifetime risk for epilepsy is about 5 %. The probability of experiencing at least one unprovoked epileptic seizure at some stage during 80 years of life is almost 1 in 15 people [4].
24.4 Clinical Features According to Classification
The electro-clinical epileptic syndromes are distinctive clinical entities made up of a complex of electro-clinical features, symptoms and signs, with a specific age of onset. Epileptic constellations, although not strictly considered syndromes, are clinical entities with distinctive features. Also for the structural/metabolic epilepsies, it is possible to link the clinical features and course to the specific underlying cause. By contrast, for the epilepsies of unknown cause, clinical features may greatly vary, and parameters dictating the clinical course and outcome are frequently difficult to establish. In the following, we will briefly delineate the clinical features of the more frequent pediatric or adult-onset epileptic forms.
24.4.1 Epileptic Encephalopathies in Newborns and Infants
Syndrome | Age at onset | Seizure type | Etiology | EEG | Neurological outcome | Epileptologic outcome |
|---|---|---|---|---|---|---|
Ohtahara | 1st month | polymorphic | congenital cortical malformations | Suppression burst | Death in 50 % of cases in the first year, severe psychomotor delay in the others | Intractable epilepsy. Possible evolution into West syndrome |
West | 4–8 months | spasms | congenital cortical malformations, tuberous sclerosis, perinatal hypoxic lesions, cryptogenic | Hypsarrythmia | Death in 5–20 %. Psychomotor regression, mental retardation | Refractory epilepsy in 95 % of cases. Possible evolution into Lennox- Gastaut syndrome. |
Dravet syndrome (severe myoclonic epilepsy in infancy) | 1st year of life | Clonic, myoclonic and partial seizures | Genetically determined mutation of the X-linked PCDH19 gene, Xq22 locus, or de novo mutation of the SCN1A gene | Rapid generalized spikes or polyspikes, photosensitivity, multifocal spikes and spike and waves. | Psychomotor regression with ataxia and myoclonus | Refractory epilepsy |
24.4.2 Focal and Generalized Epileptic Syndromes of Childhood
Syndrome | Age at onset | Seizure type | Etiology | EEG | Neurological outcome | Epileptologic outcome |
|---|---|---|---|---|---|---|
Lennox Gastaut | 3–5 yrs | Tonic and atonic with falls, atypical absences | Congenital cortical malformations, tuberous sclerosis, perinatal hypoxic lesions, cryptogenic | Slow spike and waves, polyspikes | Severe mental retardation | Refractory epilepsy |
FS+ | >6 yrs | Persistence of febrile seizures, plus afebrile generalized seizures, absences, myoclonic and focal seizures | Genetic with autosomal dominance with incomplete dominance | Not specific, with variable epileptiform abnormalities | Usually normal | Controlled seizures in most cases |
BECT | 7–10 yrs | Morpheic focal sensitive-motor with possible secondary generalization | idiopathic | Normal organization of wakefulness and sleep. Typical monomorphic central or centrotemporal SW, unilateral or bilateral, often asynchronous. | Normal neurological development | Spontaneous remission, even without treatment, in 2–4 years after onset |
Panayiotopoulos syndrome | 1–15 yrs | Autonomic signs and symptoms, frequent headache and vomiting. In 50 % of cases status epilepticus | Idiopathic | Normal organization of wakefulness and sleep. Typical SW localized most on the occipital regions. | Normal neurological development | Spontaneous remission, even without treatment, in adolescence or shortly after onset |
Late-onset childhood occipital epilepsy, or Gastaut syndrome | 8–11 yrs | Stereotypical visual auras, sometimes followed by loss of consciousness and motor phenomena | Probably genetic | Normal background, frequent occipital spikes, mainly occurring upon eye closure | Normal neurological development | Spontaneous remission in 2–7 years from onset in 50–80 % of cases. EEG abnormalities may persist |
NFLE | 1–14 yrs | Clusters of brief nocturnal motor seizures with hyperkinetic or tonic manifestations | Genetic, in most cases related to mutations of genes encoding subunits of the neuronal nicotinic acetylcholine receptor (CHRNA4, CHRNA2, CHRNB2) | Normal background, frequently normal interictal EEG; frontal low-voltage ictal spikes | Normal psychomotor development and neurologic examination in most cases | Seizure severity vary within families; seizures usually persist through adult life |
ESES | 4.5–14 yrs | Focal motor, tonic, atonic, atypical absences | Cryptogenic, or associated with heterogenic brain lesions | Multifocal and diffuse spikes and SW, that are continuous during slow sleep, occupying more than 85 % of it. Absent during REM sleep. | Progressive complex and severe neurological impairment, mainly concerning language function, with mental impairment and psychiatric disturbances | Seizures can be rather easily controlled, ESES is much more difficult to suppress, depending on the neuropsychological and behavioral outcome |
CAE | 3–12 yrs | High frequency more than daily absences, with associated eyelid myoclonia, simple automatisms | Genetic | Bilaterally synchronous 3 Hz spike and waves and interictal ictal discharges | Normal development | Good outcome, usually with complete remission, in one third of cases generalized easily controlled seizures |
Epilepsy with myoclonic absences | 2–12 yrs | Absences with bilateral rhythmical clonic jerks correlated with epileptiform EEG discharges | Genetic | Bilaterally synchronous 3 Hz spike and waves with poligraphic recording of associated myoclonia | Developmental cognitive impairment may be observed | Rather good outcome, with rare evolution toward a secondary generalized epilepsy |
Epilepsy with myoclonic-atonic seizures | 7 months- 5 yrs | Febrile and afebrile generalized seizures, followed by myoclonic-atonic seizures, determining falls, and absences | Probably genetic | Slowing of background activity, generalized irregular polyspikes and waves | Mild or even severe cognitive impairment or dementia might manifest | Rather good outcome, with tendency to relapse, mostly generalized tonic-clonic seizures |
EMA or Jeavons syndrome | 2–5 yrs | Typical absences associated with rapid eyelid myoclonia and upward deviation of the eyes | Probably genetic | Normal background, typical marked photosensitivity | Normal development | Absence seizures tend to persist into adult life |
Landau–Kleffner syndrome | 5–7 yrs | Rare focal motor and generalized, present in 70 % of patients. Progressive aphasia and psychomotor disturbances | Cryptogenic, or associated with heterogenic brain lesions | Multifocal bilateral spikes and spike and waves, often prevalent on temporal and parieto-occipital regions, activated during sleep, when they become subcontinuous | Language recovery depends on the reeducation and the correction of EEG activity | Epilepsy has a good outcome |
24.4.3 Genetic or Presumably Genetic Generalized Epilepsies of Adolescence
1.
Juvenile absence epilepsy or JAE occurs around puberty with absence seizures similar but by far less frequent than those of CAE. The incidence and prevalence of JAE in the general population are not known. Age of onset is 10–12 years.
Diagnostic markers – The EEG counterpart is a bilaterally synchronous 3–4 Hz spike and wave discharge. Generalized tonic–clonic seizures are associated in 83 % of cases, and myoclonic jerks, mainly at awakening, may be present.
Prognosis – Response to treatment for absences is very good but outcome is less favorable: JAE patients, particularly if experiencing tonic–clonic seizures, might require lifetime treatment.
2.
Juvenile myoclonic epilepsy or JME is the most common form of genetic generalized epilepsy, accounting for 5–10 % of all epilepsy cases. Age of onset is between 12 and 18 years.
Diagnostic markers – Clinical features include (i) myoclonic seizures, single or repetitive myoclonic jerks without loss of contact involving primarily upper arms and mainly occurring upon awakening; (ii) generalized tonic–clonic seizures, usually occurring a few years after myoclonic seizures, mainly on awakening, sometimes preceded by repetitive myoclonic jerks; (iii) absence seizures, in about 35 % of patients. Sleep deprivation and fatigue can facilitate seizure occurrence. The interictal EEG is abnormal in almost all patients, particularly after sleep deprivation, with 4–6 Hz poly-spike and wave discharges, which could be also asymmetric. Recent neurophysiological studies suggested that discharges in JME might involve rather restricted cortical networks including different regions of the frontal and temporal lobes.
3.
Epilepsy with generalized tonic–clonic seizures alone may occur between 6 and 20 years of age.
Diagnostic markers – It is characterized mostly by generalized tonic–clonic seizures occurring shortly after awakening, easily precipitated by sleep deprivation. Similar to that observed in JME, absence and myoclonic seizures may be present. As for JME, similar interictal poly-spike and wave discharges characterize the EEG of patients. The frequency of occurrence of this syndrome is difficult to establish, possibly for the genetic overlap with CAE, JAE, and JME.
4.
Familial TLE. A number of familial temporal lobe epilepsies have been described in the last 15 years, including familial mesial TLE [5].
Diagnostic markers – They are characterized by onset in adolescence or adulthood, benign course with medication-responsive focal seizures, and complex, possibly polygenic, mode of inheritance. Autosomal dominant epilepsy with auditory features are characterized by auditory auras preceding focal and generalized seizures, and are related to mutations of the LGI1 gene, encoding a protein associated to the voltage-gated potassium channel complex [6].
24.4.4 Progressive Myoclonic Epilepsies (PMEs)
PMEs are a heterogeneous group of progressive diseases characterized by (i) myoclonus, (ii) tonic–clonic seizures, and (iii) neurologic deterioration. Disease onset is in late childhood or adolescence.
Diagnostic markers
– In all forms, myoclonus is usually precipitated by action and posture. Causative genes have been identified for most forms. Different clinical entities are known, including Unverricht–Lundborg and Lafora diseases (the most frequent forms, see below), type III Gaucher diseases, GM2-gangliosidosis, sialidosis, some mitochondrial disorders (such as MERRF and MELAS), DRPLA, and the vast families of ceroid lipofuscinosis.
Prognosis
– Neurologic deterioration may differ greatly, from mild deficits to severe impairment and disability, in many cases progressing to death. Dementia is present in some of these disorders.
1.
Unverricht–Lundborg disease is the mildest PME form.
Diagnostic markers – In contrast to other PME, it is progressive only in adolescence. Jerks typically consist of action myoclonus triggered by voluntary movement, posture, and external stimulations. The causative disease gene is EPM1, causing massive down-regulation of cystatin B.
EEG shows normal background activity and generalized spike-waves triggered by photic driving, posterior low-amplitude spikes, and photosensitivity. By adulthood, the clinical picture becomes stable, with severe myoclonus, controlled seizures, and nearly normal cognitive functions.
Prognosis – The disease usually takes a long course, possibly with a normal life span.
2.
Lafora disease (from the Spanish neurologist Gonzalo Rodriguez-Lafora, who first described the disease and the typical accumulation bodies in neurons) is genetically related to autosomal recessively inherited mutations in the EPM2A (encoding laforin, 45 %), or EPM2B (encoding malin ligase, 45 %), or a yet undiscovered gene (remaining 10 % of cases).
Diagnostic markers – Difficulties in school, myoclonic jerks, generalized seizures and visual hallucinations are the first symptoms. EEG shows progressive slowing of background activity, posterior low-amplitude spikes, and photosensitivity, and diffuse spike and wave discharges in more advanced disease stages.
Prognosis – Myoclonus and seizures gradually worsen and become intractable, associated with dementia. Death occurs in about 6–10 years after onset, mainly due to aspiration pneumonia during status epilepticus, but some patients present a less severe form with extended survival.
24.4.5 Epileptic Constellations
They are clinical entities characterized by distinctive lesions, not affecting a specific age, but presenting with rather repeatable clinical, radiologic, and epileptologic patterns.
(a)
Diagnostic markers – Mesial temporal lobe epilepsy associated with MTS is characterized at MRI by volume reduction/atrophy and increased T2 signal of the hippocampus and parahippocampal area and, neuropathologically, by severe neuronal cell loss and gliosis of Cornu Ammonis, subfield 1 (CA1), and subiculum. It presents distinctive electro-clinical features, it is frequently drug-resistant, and it frequently leads to epilepsy surgery.
Prognosis – It is one of the most frequent medically refractory forms of human epilepsy, and the relation between MTS and drug resistance is further underscored by its presence in 50–90 % of patients operated for intractable mesial temporal lobe epilepsy [7].
(b)
Hypothalamic hamartomas (or other hypothalamic lesions) are typically associated with gelastic seizures and a particular electro-clinical pattern. Surgery is the treatment of choice (hypothalamic disconnection, but see also below).
(c)
Rasmussen Syndrome, or Rasmussen encephalitis, is a rare, inflammatory, possibly immuno-mediated chronic and progressive disease typically affecting one hemisphere [8].Diagnostic markers – Main symptoms are progressive neurological deficits, including cognitive and motor deterioration, and intractable seizures, often in the form of epilepsia partialis continua and recurring epileptic status.
Prognosis – AED treatment, long-term immunotherapy and, above all, surgery (hemispherotomy, functional hemispherectomy) represent possible therapeutic options.
24.4.6 Structural/Metabolic Epilepsies
This group of epilepsies is much more heterogenous with respect to previous ones, encompassing various types of epilepsy symptomatic of different diseases: we recognize malformations of cortical development, tuberous sclerosis, cerebral tumors, either of low or of high grade, cerebral arteriovenous malformations, cavernous angiomas, vascular lesions, such as hemorrhagic or ischemic brain lesions, and traumatic brain injuries. These underlying lesions have different intrinsic epileptogenic potential. Seizure semiology depends on the site of the epileptic focus (in general, close to but not necessarily corresponding to the lesion itself): for instance, frontal seizures with clinical frontal semiology and ictal frontal EEG pattern may be observed either in a frontal cavernous angioma, or in a frontal post-traumatic malacic lesion. Seizure semiology must guide in evaluating the lesion site, and in particular cases, the opportunity for presurgical studies.
(a)
Brain malformations or MCDs are a class of clinical entities originating from disruptions of different nature of the normal process of brain ontogenesis during prenatal life. When severe or diffuse, they represent a fairly common cause of developmental delay particularly in pediatric patients; when more localized, they are a fairly common cause of epilepsy in adolescents and adults. From a practical or clinical standpoint, the more frequent forms are lissencephaly/band heterotopia, polymicrogyria (Fig. 24.1), focal cortical dysplasia (FCD) and periventricular nodular heterotopia (PNH, Fig. 24.2). FCDs, particularly Taylor’s type FCD, are highly epileptogenic lesions, as also demonstrated by stereo-EEG (SEEG) recordings within the lesion, very frequently, but not always, associated with early-onset, drug-resistant severe epilepsy [9].
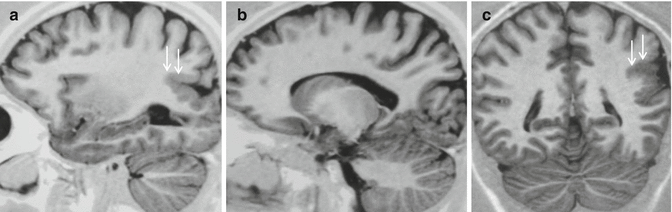
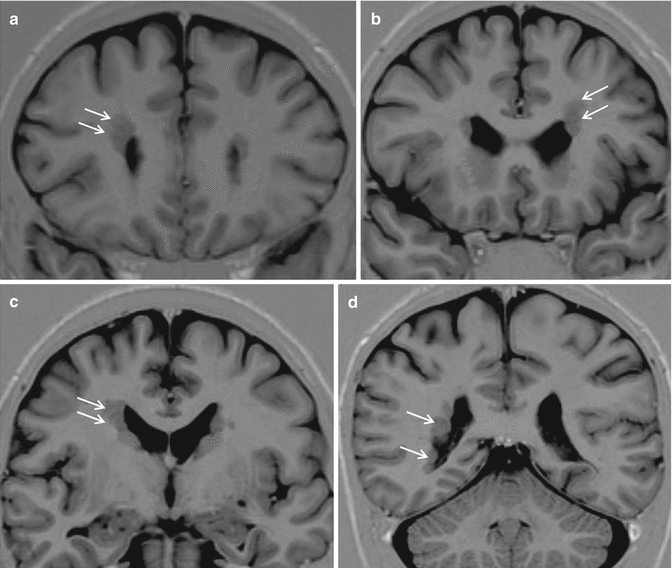
Fig. 24.1
Polimicrogyria. Sagittal and coronal IR images, a–c, demonstrate unilateral polimicrogyria involving the parietal and occipital regions (arrows): the cortical mantle is thickened, resulting from fusion of multiple adjacent small gyri. The gray-white matter interface is irregular due to fine interdigitation from abnormal convolutions
Fig. 24.2
Periventricular nodular heterotopia (PNH). Coronal IR images from anterior to posterior, a–d, show bilateral and asymmetrical multiple nodules lining the extension of the lateral walls of the lateral ventricles (arrows)
Resective surgery is the treatment of choice and is highly successful in the majority of cases, particularly in children. All PNH patients are affected by focal epilepsy, non-particularly severe but drug-resistant in the vast majority of cases. Some bilateral and symmetrical cases are genetically determined by FLN1 gene mutations. Ictal EEG and Stereo-EEG recordings in PNH patients suggest that seizures are generated by abnormal anatomic circuitries including the heterotopic nodules and adjacent cortical areas [10].
(b)
Although any tumor type can cause seizures, low-grade tumors are more frequently associated with epilepsy than high-grade tumors (e.g., high-grade astrocytomas) [11]. High-grade tumors presenting with seizures are usually smaller than high-grade tumors presenting with focal neurological deficits. Low-grade tumors, as gangliogliomas, or lesions with intermediate features between dysplasia and tumors, such as dysembryoplastic neuroepithetial tumors (DNET), present with seizures in almost 100 % of cases. Meningiomas have a much lower incidence of associated epilepsy, usually well controlled by AEDs (see Chap. 18).
(c)
Among vascular malformations, cavernous angiomas have a high incidence of epilepsy, near 99 %, largely determined by slight blood leakage and the subsequent deposit of a hemosiderin ring. Familial cases have been described, linked to mutations of three different genes (CCM1, CCM2, and CCM3). Surgery can be curative in a large percentage of cases. However, multiple cavernous angiomas may be present in the same patient, and careful MRI analysis is mandatory.
24.4.7 Epilepsies of Unknown Causes
This is a large group of epilepsies, once termed “cryptogenic,” in which a causative factor has not been found. As for the structural epilepsies (formerly called symptomatic), they can originate from the different cerebral lobes and, accordingly, when the seizure origin is sufficiently clear, they can be subdivided into frontal (mesial frontal, dorsolateral frontal, orbitofrontal and insular), temporal (neocortical and mesial), parietal, and occipital epilepsies. TLE is the most common form of adult focal epilepsy, and mesial (i.e., hippocampal) onset probably accounts for 80 % of all temporal lobe seizures [12]. Frontal lobe epilepsies are the second most common, accounting for approximately 20 % of cases. Parietal and occipital lobe epilepsies are less common, accounting for 6 % and 5–10 %, respectively, of localization-related epilepsies. The diagnostic course and treatment do not differ from those of localization-related symptomatic epilepsies.
24.5 Diagnostic Markers
For the intrinsic nature of seizures (sudden, repetitive phenomena, mostly occurring in otherwise healthy subjects), in most cases the diagnosis of epilepsy largely relies on the accurate evaluation of the clinical history of each individual patient.
Laboratory tests (serum and CSF)
– In general, laboratory tests in epilepsy take on more relevance in the routine management of patients rather than in the diagnostic procedure. Indeed, routine blood and urine tests are mandatory to monitor possible side effects of the different AEDs on blood cell production, hepatic and renal function, and electrolyte balance. Regular blood sampling is also fundamental for monitoring basal of almost all AEDs (in the morning, before taking the first dose of the day). For a detailed analysis of the proper use of therapeutic drug monitoring see [13].
CSF analyses
are obviously of pivotal importance in the diagnosis of systemic and CNS infections (e.g., viral, bacterial, fungal, and parasitic meningitis or meningo-encephalitis). More recently, it has been increasingly recognized that some epilepsy-related encephalitis can be antibody-mediated. Antibodies to voltage-gated potassium channels can provoke limbic encephalitis, characterized by focal seizures of temporal origin, amnesia, and unilateral or bilateral mesial temporal lobe inflammation possibly evolving into atrophy [14]. Antibodies to NMDA receptor subunits may provoke, predominantly in young women, a subacute-onset encephalopathy characterized by psychic symptoms, movement disorders, and focal seizures [15]. Other auto-antibodies possibly involved in the pathogenesis of focal epilepsy (particularly in temporal lobe epilepsy) include antibodies against two proteins associated to the voltage-gated potassium channel complex, CASPR2 and LGI1, anti-GABA receptors, GAD, and AMPA receptor subunits. When the patient clinical history is suggestive of an acute- or subacute-onset encephalopathy with seizures, these antibodies should be searched for in both CSF and serum.
Imaging
– CT scan is of little help, except for recognizing calcifications in very restricted cases. MRI is mandatory, with contrast medium if a tumor or a lesion determing blood-brain-barrier disruption is suspected. MRI standardized protocols can now easily identify different epileptogenic lesions (e.g. MTS in the temporal lobes). However, it is always wise not to make an etiological diagnosis based on imaging only, since the finding of a brain lesion can be casual, and the epileptic focus can be localized at distance, or extend outside the boundaries of a lesion. Lesions can be judged as the cause of epilepsy if a good correlation exists with the electro-clinical data, i.e., semiologic ictal signs and symptoms, and interictal as well as ictal EEG. This notion is of particular relevance in the planning of a surgical strategy.
PET is another useful tool in detecting an area of reduced fluorodeoxyglucose uptake, usually related to the site of epileptic focus, even in presence of apparent structural normality. SPECT can give information on seizure focus, and can be employed in ictal (hyper-perfusion) and interictal (hypo-perfusion) conditions.
Genetics
– Recently, a number of genes have been reported as causative for several epilepsy syndromes (mutations in the ARX, CDKL5, SLC25A22, and STXBP1 genes in Ohtahara syndrome; ARX, CDKL5, SLC25A22, SPTAN1, PLCb1, MAGI2, and PNKP gene mutations in West syndrome. SCN1B, SCN1A, SCN2A, SCN9a, GABRG2, GABRD are genes mutated in families with febrile seizures plus; SCN1A and PCDH19 gene mutation in Dravet syndrome; mutations of genes encoding subunits of the neuronal nicotinic acetylcholine receptor, CHRNA4, A2, and B2, in most cases of autosomal dominant nocturnal frontal lobe epilepsy; LGI1 mutations in autosomal dominant epilepsy with auditory features). In addition, causative genes are known for most cases of progressive myoclonic epilepsies, such as mutations of EPM1 for Unverricht–Lundborg, EPM2A and EPM2B for Lafora, PolG1 and mitochondrial DNA for MERRF and MELAS. Mutations have been found in some cases of malformations of cortical development such as of the FLN1 gene for PNH, DCX and LIS1 for lissencephaly/double cortex syndrome, different genes for various types of familial polymicrogyria, and the DEPDC5 gene for familial patients with focal cortical dysplasia [16]. Mutations of the CCM1, CCM2, and CCM3 genes have been found in familial but also sporadic cases of epileptogenic cavernous angiomas [17].
It is likely that a considerable degree of genetic overlap exists between CAE, JAE, JME, and epilepsy with tonic–clonic seizures alone. For CAE and JAE, a polygenic mode of transmission is the most likely mode of inheritance. Susceptibility to gene mutations in different ion channels or linkage for other genetic regions has been reported. For JME, different mutations can determine the same clinical phenotype. Both autosomal dominant (for the GABRA1 gene) and recessive inheritance have been reported in different families. As for CAE and JAE, the most likely mode of inheritance is polygenic transmission.
In pediatric focal epilepsies, the typical EEG trait of BECT could be dictated by an autosomal dominant pattern of inheritance. However, the combination of other genetic or even extrinsic influences may be relevant for the clinical expression of BECT (brain structural lesion have been also associated to BECT). A genetic predisposition is also likely for Panayiotopoulos and Gastaut syndrome, since a family history for epilepsy is present in 20–30 % of affected patients. In a severe and early-onset Panayiotopoulos patient, a mutation of the SCN1A gene was reported, whereas no linkage to specific chromosome regions has been demonstrated so far for Gastaut syndrome.
A more detailed update of genes relevant for the different epilepsy syndromes can be found at the LICE website, Genetic Commission, at www.lice.it.
Neurophysiology
Interictal scalp EEG during wakefulness and sleep can provide relevant support to the clinical diagnosis of epileptic seizures. By no means does a normal EEG exclude a diagnosis of epilepsy. Ictal EEG scalp recordings are useful in confirming the diagnosis and in the differential diagnosis with non-epileptic phenomena. EEG monitoring is mandatory for epilepsy presurgical studies, in which the site of epileptic focus should be determined to decide opportunity and strategy of surgery.
A rather new neurophysiological method, magnetoencephalography or MEG offers interesting possibilities to be verified in the near future. MEG analyzes interictal EEG abnormalities, with much better spatial resolution than surface EEG, particularly when abnormal activity is generated in a cortical sulcus. MEG data can be projected on specific MRI images, thus providing relevant information on the location of the epileptic focus.
24.6 Special Situations
24.6.1 Febrile Seizures
They are generalized, age-related seizures, occurring during an acute febrile illness, in children aged from 6 months to 5–6 years. About 3 % of the population experiences febrile convulsions. They can occur in children with normal development, be simple and of short duration. Early (within the first year of life), repeated, lateralized, and prolonged febrile convulsions, and the existence of familiarity for epilepsy are all factors more likely associated with later epilepsy development (see below, prognosis).
24.6.2 The First Seizure
About 6 % of the population will experience a single afebrile seizure in life. In the case of provoked seizures (i.e., related to electrolyte imbalance, alcohol abuse or withdrawal, and precipitating medications), they are not diagnosed within the frame of epilepsy and AED treatment is not required.
24.7 Prognosis
Several prognostic factors for recurrence have been identified (although not without controversy), including congenital neurological deficits, EEG epileptiform abnormalities, family history for epilepsy, and the presence of focal and nocturnal seizures, or mixed seizure types. However, the timing of treatment at the onset of epilepsy has no effect on long-term prognosis. Different studies demonstrated that even if patients treated after the first unprovoked seizure had a reduced risk of recurrence after 24 months compared with untreated patients, the probability of reaching 1 or 2 years remission was the same when therapy was started after the first or the second seizure [18].
24.7.1 Status Epilepticus (SE)
24.7.1.1 Definition
SE is defined as an epileptic activity continuing for 30 min or more or a series of epileptic seizures during which function is not regained between seizures for longer than 30 min.
Non-convulsive types of SE include:
(a)
Absence SE and myoclonic SE, occurring in idiopathic generalized epilepsy of childhood and adolescence, but also rarely in adult or even elderly patients, as a result of drug withdrawal;
(b)
Complex partial SE, more frequently occurring in adults; and
(c)
Epilepsia partialis continua, characterized by repetitive focal motor seizures recurring every few seconds or minutes for very prolonged periods of time.
Convulsive or tonic–clonic SE is the more dangerous form, since it may evolve into refractory SE (when treatment with anesthetics is required), or super refractory SE (lasting more than 24 h under anesthesia or recurring when anesthetics are reduced or withdrawn), which are characterized by high mortality rates (see below).
24.7.1.2 Epidemiology
Incidence of SE ranges from 10 to 60 cases/105 per year, according to seven population-based studies of SE, peaking in the very young and the elderly [19]. The frequency of underlying major etiologies varies among published studies and show marked geographic differences. Major causes include febrile illnesses associated with systemic and CNS infections (particularly in children), acute cerebrovascular diseases (stroke is the more prevalent cause with increasing age), alcohol and drug abuse, and metabolic disorders. Brain tumors and trauma are less frequent cause of SE. In a sizeable proportion of cases (up to 15 % of patients), no evident cause is present (cryptogenic SE). In patients with a previous history of epilepsy, SE is frequently determined by low or absent antiepileptic drug levels [19].
24.7.1.3 Treatment
The treatment protocol for tonic–clonic SE, although not sufficiently supported by controlled study on efficacy and follow-up, is widely accepted and used in most tertiary care centers [20].
In the early phases of SE rectal, buccal, intranasal (if out-of-hospital), or intravenous – IV – (if facilities for resuscitation are available) benzodiazepines are first used, followed by IV AEDs (phenytoin, fosphenytoin, phenobarbital, valproic acid) and, if necessary, by the induction of anesthesia (midazolam, thiopental or phenobarbital, and propofol [21], also see below).
Although the lack of controlled studies hampers the evaluation of literature data, midazolam seems superior to propofol and barbiturates (thiopental or pentobarbital) in terms of reduced death rates during infusion, reduced breakthrough seizures (i.e., recurring after initial control and dictating switch of therapy) and withdrawal seizures (occurring after drug withdrawal) [20]. The use of propofol can be complicated, particularly in patients co-medicated with steroid or cathecolamines or children with prolonged treatment, by the frequently fatal propofol infusion syndrome, leading to rhabdomyolysis, metabolic acidosis, and heart and renal failure. For details in the use of a variety of second-line treatments of SE, including AEDs, see [20].
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


