Epilepsy as the Presenting Manifestation of Structural Brain Lesions
Although epilepsy can be caused by a host of brain lesions of congenital or acquired origin, these causes are not reviewed in detail here; this chapter deals only with some of the organic causes of epilepsy that have been selected because of their diagnostic importance; their frequency; or the specific problems they pose regarding therapy, investigations to be performed, or genetic counseling. Selected causes include brain tumors, abnormalities of cortical development, tuberous sclerosis (TS), hippocampal sclerosis (HS), vascular malformations, Sturge-Weber syndrome, and cerebral palsy.
BRAIN TUMORS
Seizures can occur with both malignant and benign tumors, and they may be their first clinical manifestation. They may be caused by the effect of the tumor on the surrounding cortex; by associated lesions that may be contiguous to the tumor, such as cortical dysplasia (Prayson and Estes, 1995; Prayson et al., 1993), or by distant lesions involving especially the mesial temporal lobe. In the latter case (dual pathology), the respective roles of tumor and the other lesions remain a subject of dispute (Cendes et al., 1995). In rare cases, the tumor itself can be the origin of seizure discharges (Harvey et al., 1996; Munari et al., 1995).
Epileptic seizures in childhood are only seldom caused by brain tumors. In unselected series of children with epilepsy, only 0.2% to 0.3% had brain tumors (Livingston, 1972; Page et al., 1969), although a higher proportion of tumors has been found in some selected series, especially when epilepsies with partial seizures have been considered (Blume et al., 1982; Aicardi et al., 1970). Thus, Gilsanz et al. (1979) found three tumors among 169 children with apparently cryptogenic partial epilepsy (i.e., isolated seizure disorders without neurologic signs or concomitant disease). Aicardi et al. (1983) found three tumors among 274 children with apparently cryptogenic partial epilepsy without neurologic signs or concomitant disease. Hauser et al. (1993) found an incidence of 1% in patients younger than 15 years, and Sander and Sillanpää (1998) found an incidence of 1% in those younger than 30 years. Only 2% of infants younger than 1 year with tumor present with a seizure. Seizures are uncommon in children with infratentorial tumors. On the other hand, epileptic seizures are the first manifestation of tumors of the cerebral hemispheres in one-fourth (Gilles et al., 1992) to half (Aicardi et al., 1970) of the cases in both children and adults.
Most tumors that give rise to seizures are benign; thus, an early diagnosis is important because many are amenable to surgical treatment. The problem is, therefore, selecting from the extremely large number of children with seizures those few whose attacks are likely to be caused by brain tumors, while minimizing the number of patients submitted to neuroimaging diagnostic procedures. Epilepsy also may be a consequence of the treatment (surgical or otherwise) of recognized brain tumors, but such epilepsies are not dealt with in this chapter.
Any type of tumor can give rise to seizures, but benign tumors are most often the cause of isolated epilepsy. Rapidly growing tumors usually present with focal deficits and/or intracranial hypertension. Low-grade gliomas, especially astrocytomas and oligodendrogliomas and mixed oligoastrocytomas, were mostly found in early studies (Aicardi et al., 1970). More recent studies have emphasized the role of neuronal glial tumors, especially gangliogliomas and developmental neuroepithelial tumors (DNTs), in the causation of chronic epilepsy; these may remain monosymptomatic in the form of recurrent partial seizures for many years. The frequency of the different types varies from center to center, and this may depend partly on the pathologic criteria used. At the Maudsley Hospital, London, Bruton (1988) found that, of 37 tumors from 249 temporal lobectomies, nine were gangliogliomas. Of 216 temporal lobectomies
performed in Bonn, Germany, 75 contained tumors, of which 34 (45%) were gangliogliomas and 25 (33%) were astrocytomas (Wolf et al., 1993). The idea that, regardless of histologic diagnosis, indolent tumors associated with chronic epilepsy may constitute a distinct clinicopathologic group has been expressed (Fried et al., 1994). Likewise, Bartolomei et al. (1997) suggested that low-grade temporal and extratemporal tumors found in 45 patients shared many features with DNTs in terms of their clinical profile and course and that they may represent variants of DNTs (Pasquier et al., 2002). The latter are diagnosed especially in children and adolescents; they are often superficially located in the temporal lobe, and they tend to remain static over time. They often manifest as isolated partial seizures of early onset for quite long periods (Raymond et al., 1994b; Kirkpatrick et al., 1993; Daumas-Duport, 1988), although they may be associated with cognitive and behavioral deterioration in a few early cases (Neville et al., 1997). Their histologic appearance is heterogeneous, and their differential diagnosis may be difficult (Honavar et al., 1999). Gangliogliomas and gangliocytomas are closely related tumors that may occur even in neonates (Duchowny et al., 1989), and their total removal with subsequent seizure control is often feasible (Duchowny et al., 1996; Zeutner et al., 1994).
performed in Bonn, Germany, 75 contained tumors, of which 34 (45%) were gangliogliomas and 25 (33%) were astrocytomas (Wolf et al., 1993). The idea that, regardless of histologic diagnosis, indolent tumors associated with chronic epilepsy may constitute a distinct clinicopathologic group has been expressed (Fried et al., 1994). Likewise, Bartolomei et al. (1997) suggested that low-grade temporal and extratemporal tumors found in 45 patients shared many features with DNTs in terms of their clinical profile and course and that they may represent variants of DNTs (Pasquier et al., 2002). The latter are diagnosed especially in children and adolescents; they are often superficially located in the temporal lobe, and they tend to remain static over time. They often manifest as isolated partial seizures of early onset for quite long periods (Raymond et al., 1994b; Kirkpatrick et al., 1993; Daumas-Duport, 1988), although they may be associated with cognitive and behavioral deterioration in a few early cases (Neville et al., 1997). Their histologic appearance is heterogeneous, and their differential diagnosis may be difficult (Honavar et al., 1999). Gangliogliomas and gangliocytomas are closely related tumors that may occur even in neonates (Duchowny et al., 1989), and their total removal with subsequent seizure control is often feasible (Duchowny et al., 1996; Zeutner et al., 1994).
However, malignant tumors (high-grade astrocytomas) have been found in a few cases of long-standing epilepsy, which suggests that malignant changes may occur within benign tumors with time (Aicardi et al., 1970; Page et al., 1969).
Clinical and Electroencephalographic Features of Epilepsies Caused by Brain Tumors
The clinical and EEG features of the epilepsies that reveal tumors of the cerebral hemispheres in children are important to consider mainly in those patients in whom epilepsy is not immediately or very rapidly associated with other neurologic signs and symptoms or with signs of intracranial hypertension. In many patients with hemispheral tumors, epileptic seizures remain the sole clinical manifestation for a few months to many years (Spencer et al., 1984a). In one series (Aicardi et al., 1970), the mean interval between the first seizure and the diagnosis of a tumor was 5.6 years, and intervals as long as 20 years have been reported (Blume et al., 1982; Aicardi et al., 1970; Page et al., 1969; Lennox and Lennox, 1960). In the same series, 8% of patients were younger than 1 year and 19% younger than 2 years, and cases occurring in infancy or the neonatal period have been recorded (Duchowny et al., 1989; Rutledge et al., 1987). Most cases, however, occur in older children and adolescents (Kirkpatrick et al., 1993; Blume et al., 1982; Page et al., 1969). In mixed series of children and adults (Cascino, 1990; Boon et al., 1991), refractory epilepsy commonly is the sole manifestation of a tumor. Boon et al. (1991) found that 50 of 250 patients with intractable partial seizures had a detectable lesion on neuroimaging and that the lesion was neoplastic in 35 (70%). Most patients had an onset of seizures at adolescence (mean age of 13 years), and the mean duration of the epilepsy was 11 years. The lesions were located in the temporal lobe in 57% of patients. This series included four cases of hamartoma and three of ganglioglioma. Cascino (1990) and Cascino et al. (1993a) similarly reported that the seizures were refractory to drug treatment in 30 of their 45 patients. The widespread use of neuroimaging techniques has shortened the diagnostic delay considerably.
Partial seizures are more common than generalized seizures, but the latter are not uncommon. Several types of attacks are commonly associated, but generalized seizures can occur in isolation for months or years. Some seizures are described as “generalized” because of nonlocalizing symptoms, such as staring, eye blinking, generalized hypertonia, or hypotonia, are the expression of localized, often temporal discharges (Spencer et al., 1984a; Aicardi et al., 1970). Some types of seizures (e.g., complex partial seizures with olfactory hallucinations) are usually regarded as symptomatic of a tumor in a high proportion of cases (Penfield and Jasper, 1954), although this has been debated (Howe and Gibson, 1982). In the authors’ experience, purely or predominantly sensory symptoms may often be associated with central tumors. In a series of 98 children with supratentorial astroglial tumors and seizures, partial seizures were part of the presentation in half of the cases, and they were the only complaint in 30% (Shady et al., 1994).
A tumor should be readily suspected when deterioration in personality or performance becomes evident or when progressive neurologic deficits or signs and symptoms of increased intracranial pressure appear. However, especially with developmental tumors, epilepsy often remains the only abnormality for long periods lasting up to several years (Duchowny et al., 1996; Raymond et al., 1994b). These tumors are often localized to the temporal lobe, especially its mesial aspect, but they may also arise from the frontal and other lobes. They are often resistant to drug therapy, and they are not associated with any neurologic sign or other symptoms.
Contrary to what is often stated, no pattern of seizure recurrence is indicative of a tumor, and patients with hemispheral tumors may have only occasional fits or seizures that respond favorably to drug therapy, at times for prolonged periods. Neurologic signs appear late, and the correct diagnosis should not await their appearance. Behavioral and/or intellectual deterioration, on the other hand, may be more precocious (Aicardi et al., 1970; Page et al., 1969). Blume et al. (1982) found that the likelihood of tumors was highest in patients with a normal intellect.
The electroencephalographic (EEG) abnormalities associated with brain tumors have been extensively reviewed (Daly and Markand, 1990; Hughes and Zak, 1987; Blume, 1982; Bancaud et al., 1973; Page et al., 1969). No EEG pattern is specific for tumors. Localized foci of polymorphic delta waves are the most suggestive, but these are often absent. Foci of sharp waves on a slow disorganized background are almost as common, and these are suggestive of an organic lesion. Focal spiking without significant changes in background tracing is not rare. The EEG anomalies associated with developmental tumors seem more diffuse than those associated with hippocampal atrophy (Hamer et al., 1999; Blume et al., 1982). Multifocal spiking may occur (Blume et al., 1982), and it seems relatively common in children. Bilateral synchronous discharges, either asymmetric or symmetric, have been reported in 10% to 25% of patients (Page et al., 1969; Madsen and Bray, 1966). A normal EEG is rare with tumors of the cerebral hemispheres (5% of patients), but it may be seen in 10% to 20% of patients with deep midline tumors or tumors of the parasagittal region (Hughes and Zak, 1987).
Several unusual epileptic presentations can occur in children with tumors. Infantile spasms are rarely caused by brain tumors (Kotagal et al., 1995a; Ruggieri et al., 1989), and this is also true for the Lennox-Gastaut syndrome (LGS) (Angelini et al., 1979). Two uncommon syndromes deserve special attention.
Diencephalic hamartomas may be responsible for a well-defined epileptic syndrome (Berkovic et al., 1988; Breningstall, 1985; Curatolo et al., 1984; Diebler and Ponsot, 1983; Matustik et al., 1981) that is characterized by gelastic (giggling) seizures with onset in the first 2 years of life. Such giggling attacks are usually frequently repeated. They are often associated with progressive mental retardation (Berkovic et al., 1988, 2003; Deonna and Ziegler, 2000), especially when the seizures evolve into secondary generalized attacks reminiscent of those in LGS (see Chapter 4).
Another rare syndrome of epilepsy arising from deep structures has recently been reported under the terms cerebellar epilepsy (Arzimanoglou, 1999; Harvey et al., 1996) or subcortical epilepsy (Arzimanoglou et al., 1999). It is due to a hamartomatous tumor located in the cerebellar peduncle and brainstem, and it is manifested by repeated brief attacks of hemifacial contracture from early life. Harvey et al. (1996) demonstrated that the attacks were associated with a typical epileptic discharge originating in the vicinity of or within the tumor. This lesion may be amenable to surgical therapy, which may control the seizures if the resection is complete (Harvey et al., 1996) (see Chapter 10).
Neuroimaging Findings in Brain Tumors
Modern neuroimaging has resulted in a considerable change in the circumstances of the diagnosis, although some tumors continue to be missed for substantial periods. This occurs especially when only a computed tomographic (CT) scan is performed, particularly when the tumors involve the temporal lobe because of the common presence of bone artifacts in that area. Missing a tumor also can result from a failure to use contrast enhancement, which should be used routinely in this clinical context, or from the use of less than optimal machines. In the series of Spencer et al. (1984a), the initial CT scan was not diagnostic in 5 of 20 patients. Two of the CT scans were misinterpreted, but three were normal, even in retrospect, demonstrating that repeat examination may be necessary in some cases.
Magnetic resonance imaging (MRI) is clearly preferable to CT (Commission on Neuroimaging of the International League Against Epilepsy, 1997), and MRI should be the first imaging investigation performed for epilepsy. A CT scan may be used in special indications, especially for the demonstration of calcifications, which are better visualized with CT than with MRI.
Neither CT nor MRI are indicated in children with only generalized epilepsy of the idiopathic type because the yield of imaging in such patients is extremely low (Harwood-Nash, 1983; Varma et al., 1983). These include the primary generalized epilepsies of adolescence, various types of myoclonic epilepsy of late childhood, and typical absence epilepsy. Imaging is also unnecessary in typical cases of partial benign epilepsy with rolandic focus. If the physician does not feel secure enough about his or her ability to diagnose benign partial epilepsy or one of the other syndromes for which neuroradiologic examination is not indicated, imaging should be conducted. On the other hand, the clinician need not wait before
obtaining an imaging study as long as he or she is not dealing with a child who has an occasional seizure because tumors can be detected even at a very early stage. Certainly, neuroimaging has made the diagnosis of brain tumors easier at an earlier stage.
obtaining an imaging study as long as he or she is not dealing with a child who has an occasional seizure because tumors can be detected even at a very early stage. Certainly, neuroimaging has made the diagnosis of brain tumors easier at an earlier stage.
MRI has considerably improved the diagnosis of small tumors. The possibility of using different planes of imaging has considerable value for the detection of small lesions such as those in the hippocampus. Several sequences should be used, including fluid-attenuated inversion recovery (FLAIR) images, thin cuts, three-dimensional acquisition, reformatting in different planes, and gadolinium enhancement. In most cases, this should distinguish tumors from dysplastic and inflammatory lesions. However, distinguishing peritumoral edema from the tumor itself may remain difficult, thus making the delimitation of the lesion difficult. Repeating the examination after a few months can be necessary when doubt about the nature and a possible change in volume of the lesion exist.
Astrocytomas usually present as areas of increased signal, often with cystic formation (Fig. 19.1). Oligodendrogliomas involve the white matter, with possible infiltration of the neighboring cortex. Gangliogliomas may not show well on T1-weighted sequences, but they do appear as an area of increased signal that usually involves both the gray and the white matter (Duchowny et al., 1996; Sutton et al., 1983).
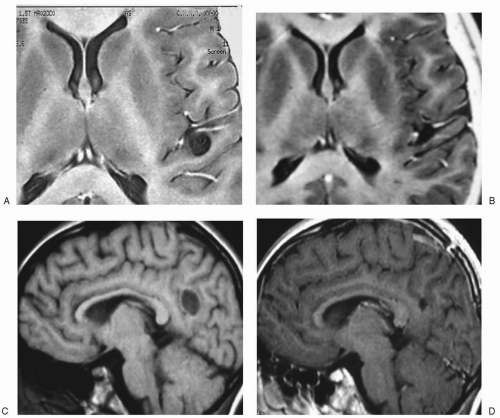 FIG. 19.1. Preoperative (A) and postoperative (B) magnetic resonance imaging (MRI) scan of a 7-year-old boy who presented with partial motor seizures related to the presence of an astrocytoma. He has been seizure free since surgery. Preoperative (C) and postoperative (D) MRI scans of an astrocytoma responsible for gyratory seizures in a 12-year-old girl. (Courtesy of Pr. Sainte-Rose, Hospital Necker, Paris, France.) |
Dysembryoplastic neuroepithelial tumors are well demarcated, often multilocular, lesions that may be cystic and/or calcified (Figs. 19.2 and 19.3). They mostly involve the cortex, although they may be associated with edema of the white matter or they may impinge on it. They may be found in any lobe, but they most often involve the mesial or external aspects of the temporal lobe. They may enhance with gadolinium, and, occasionally, they may be visible after only contrast enhancement (Raymond et al., 1994b; Pasquier et al., 2002). When they are located near the inner table of the skull, they usually expand the overlying calvarium.
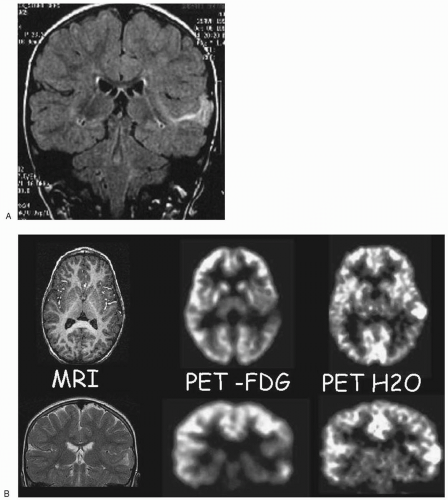 FIG. 19.2. A, B: Histologically confirmed dysembryoplastic neuroepithelial tumor (DNET) (magnetic resonance imaging and positron emission tomography), localized over the left posterior temporal gyrus, in a 2.5-year-old boy who presented with an episode of status followed by aphasia. No seizures were observed during the following 12 months. However, aphasia persisted despite intensive speech therapy. A left temporal spike-wave focus was present in all of the interictal electroencephalograms. Furthermore, long-duration recordings evidenced sequences of attenuation of the focus that were followed by rhythmic theta activity of low amplitude and flattening without any accompanying clinical signs. Two months later, clinical seizures were observed again. He was operated on at the age of 4 years (partial gyrectomy with complete excision of the DNET), and he has been seizure free for the last 18 months. His language acquisition has rapidly progressed back to normal. |
Finally, attention should be paid to the detection of other lesions, the so-called dual pathology (Cendes et al., 1995; Raymond et al., 1994a; Levesque et al., 1991).
The second lesion is usually HS, but other abnormalities are possible. Cendes et al. (1995) found hippocampal atrophy in 23 (14%) of 167 patients (both adults and children) with partial seizures that was mostly temporal. The frequency of dual pathology in this series was 2% in tumors, as opposed to 9% in vascular malformations and 25% in neuronal migration defects. Although the precise significance of dual pathology is still unclear, it should be detected and its role discussed when a surgical operation is being contemplated.
The second lesion is usually HS, but other abnormalities are possible. Cendes et al. (1995) found hippocampal atrophy in 23 (14%) of 167 patients (both adults and children) with partial seizures that was mostly temporal. The frequency of dual pathology in this series was 2% in tumors, as opposed to 9% in vascular malformations and 25% in neuronal migration defects. Although the precise significance of dual pathology is still unclear, it should be detected and its role discussed when a surgical operation is being contemplated.
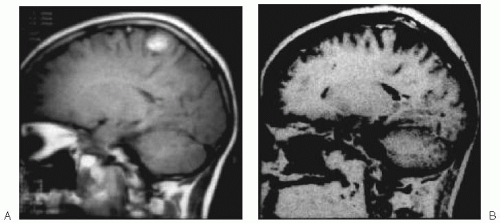 FIG. 19.3. Dysembryoplastic neuroepithelial tumor in a 9-year-old girl who presented with episodes of twitching and contraction of the right calf. The episodes could be associated with hypoesthesia of the leg, difficulty controlling movement, or even postictal paresis. A neurologic examination showed no abnormalities. After surgery (right), she remained seizure free. (Courtesy of Dr. Philippe Kahane, Grenoble University Hospital, France.) |
The common practice of imaging has uncovered some new diagnostic problems, especially those raised by the discovery of localized, more or less rounded areas of signal change, which may or may not represent indolent gliomas. When doubt persists, repeated imaging or biopsy of the lesion may be indicated. A signal change on MRI may be due to changes in blood-brain barrier permeability following seizures (Yaffe et al., 1995), and the investigations should resolve whether this is due to brain edema, hence emphasizing the need for follow-up examinations in cases of doubt.
Treatment
The treatment of epilepsy caused by brain tumors consists primarily of treating the tumor when it is clearly an aggressive, growing lesion. When the epilepsy is isolated, the treatment of seizures begins with antiepileptic agents, followed by treatment of the underlying tumor, usually by surgical resection. In most cases of indolent tumors, especially in dysembryoplastic neuroepithelial tumors and similarly behaving tumors, no other treatment is indicated because these are benign lesions that seldom, if ever, raise oncologic problems. However, not all tumors, including indolent ones, are always resectable; in such cases, partial resection may be successful. Monitoring of the lesion by follow-up MRI is probably acceptable when the prospect of resection is poor (Duchowny et al., 1998).
Excellent results have been reported after complete resection of the lesion, especially small indolent tumors, without specific attempts at the removal of the epileptogenic area (Boon et al., 1991; Cascino, 1990; Hirsch et al., 1989; Drake et al., 1987). In a large series of children with small glial tumors, Hirsch et al. (1989) reported control of epilepsy in 86% of their patients. The results were particularly satisfactory for those patients with the more benign pathologies in temporal, rather than extratemporal, epilepsy (Cascino et al., 1993a). In a series of 49 patients with alien tissue lesions, Bruton (1988) observed complete relief in 4 of 5 patients with a ganglioglioma and in 10 of 11 of those with mixed glial lesions, some of which would probably now be considered dysembryoplastic neuroepithelial tumors. Similar results were reported by Kirkpatrick et al. (1993).
Whether lesionectomy alone represents the optimal therapy remains a topic of debate. The relationship between a tumor and the origin of epileptic seizures is complex (Bancaud et al., 1973). Epileptic activity may not arise within the tumor itself, which is electrically silent when depth electrodes are inserted into it; rather, it may originate in the modified surrounding
neural tissue (Engel, 1982a; Talairach and Bancaud, 1974; Falconer and Cavanagh, 1959). The question arises, therefore, of the removal of both the tumor tissue and the epileptogenic area that have been previously located by neurophysiologic studies (Cascino, 1990; Taft and Cohen, 1971). Some investigators think complete tumor resection, including additional resection of the epileptogenic area as determined by electrophysiologic investigation, electrocorticography (EcoG), or depth electrodes, is more effective. Cascino et al. (1992c) treated 23 patients by computer-assisted lesionectomy; 17 of them (74%) had a greater than 90% reduction in seizures, and 13 (56%) had an Engel grade 1 outcome. Boon et al. (1991) studied 50 patients (38 with tumors) who had a lesionectomy only; 83% were seizure free, and 11% had a greater than 90% reduction of seizures. Berger et al. (1991) used EcoG to achieve complete resection of the epileptogenic area in 45 adult patients with glial tumors; 41 (91%) became seizure free. Jooma et al. (1995) treated adults with lesionectomy, with only 3 patients becoming seizure free, whereas, of the 14 who had lesionectomy plus resection of the epileptogenic area as defined by corticography, 13 became seizure free. Montes et al. (1995) used lesionectomy alone in 18 children, 16 of whom (89%) were rendered seizure free. Although the current data are difficult to interpret because of methodologic problems, the resection of the epileptogenic area appears logical, especially because certain tumors (dysembryoplastic ones) are often associated with areas of dysplasia that may well be epileptogenic (Raymond et al., 1994b; Prayson et al., 1993).
neural tissue (Engel, 1982a; Talairach and Bancaud, 1974; Falconer and Cavanagh, 1959). The question arises, therefore, of the removal of both the tumor tissue and the epileptogenic area that have been previously located by neurophysiologic studies (Cascino, 1990; Taft and Cohen, 1971). Some investigators think complete tumor resection, including additional resection of the epileptogenic area as determined by electrophysiologic investigation, electrocorticography (EcoG), or depth electrodes, is more effective. Cascino et al. (1992c) treated 23 patients by computer-assisted lesionectomy; 17 of them (74%) had a greater than 90% reduction in seizures, and 13 (56%) had an Engel grade 1 outcome. Boon et al. (1991) studied 50 patients (38 with tumors) who had a lesionectomy only; 83% were seizure free, and 11% had a greater than 90% reduction of seizures. Berger et al. (1991) used EcoG to achieve complete resection of the epileptogenic area in 45 adult patients with glial tumors; 41 (91%) became seizure free. Jooma et al. (1995) treated adults with lesionectomy, with only 3 patients becoming seizure free, whereas, of the 14 who had lesionectomy plus resection of the epileptogenic area as defined by corticography, 13 became seizure free. Montes et al. (1995) used lesionectomy alone in 18 children, 16 of whom (89%) were rendered seizure free. Although the current data are difficult to interpret because of methodologic problems, the resection of the epileptogenic area appears logical, especially because certain tumors (dysembryoplastic ones) are often associated with areas of dysplasia that may well be epileptogenic (Raymond et al., 1994b; Prayson et al., 1993).
Overall, the best outcome for seizure relief is when the tumor can be excised completely. The prognosis can probably be improved if the epileptogenic cortex defined by electrophysiologic studies is removed with the tumor. However, even partial resection may, in some cases, give good seizure control.
ABNORMALITIES OF CORTICAL DEVELOPMENT
Abnormal cortical development is increasingly recognized as a cause of both epilepsy and developmental disabilities (Vigevano et al., 2003). Histologically proven developmental brain abnormalities are observed in up to 25% of children with intractable seizures (Kuzniecky and Powers, 1993). Most such abnormalities may now be detected with MRI. However, some cortical malformations, or cortical dysplasias, remain undetectable even with the best imaging techniques. Although such information can detect even very slight structural alterations, it does not always provide a precise indication of the pathologic nature of the lesion. The use of generic terms, such as dysplasias (Robain, 1996) or malformations of cortical development (Barkovich et al., 2001) or of the cerebral cortex, appears more appropriate whenever the exact nature of the developmental lesion is unclear on MRI and pathologic studies are unavailable. More specific terms may be used with highly characteristic MRI patterns (Barkovich, 1996).
Abnormalities of the cerebral cortex may be diffuse, or they may involve discrete cortical areas. The development of the cerebral cortex involves three distinct, but overlapping, processes, including neuronal and later glial proliferation, neuronal migration, and cortical organization. Cortical malformations can originate from abnormalities of any or all of these processes. Often, classifying cortical dysplasias according to their supposed mechanism is difficult because some dysplasias (e.g., polymicrogyria) may represent distinct histologic types of abnormalities and because different pathologic processes can coexist in the same macroscopic lesion (e.g., heterotopia and polymicrogyria). In fact, defects in one mechanism (e.g., differentiation) can cause secondary disturbances because the abnormal cells do not migrate or differentiate normally.
Certain malformations are genetically determined (Guerrini and Carrozzo, 2001a), and, for others, a genetic origin has been hypothesized. Some may be linked to prenatal insults (Sarnat, 1992; Friede, 1989). In most cases, however, the cause remains unknown. Some malformations are more epileptogenic than others. In specific forms, the epileptogenesis appears to originate from the intrinsic properties of the dysplastic tissue (Guerrini et al., 1999a; Mattia et al., 1995). The following sections review the most common disorders of cortical development and the characteristics of the associated epilepsy and EEG patterns.
Anomalies Related to Abnormal Proliferation and Differentiation of Neurons and Glia
Hemimegalencephaly
In hemimegalencephaly (HME), one cerebral hemisphere is enlarged and structurally abnormal, with a thick cortex, wide convolutions, and reduced sulci. The abnormality is strictly unilateral (Robain and Gelot, 1996). Laminar organization is absent in the cortex, and the demarcation between gray and white matter is poor. Giant neurons (up to 80 µ in diameter)
are found throughout the cortex and the underlying white matter. In about 50% of patients, large bizarre cells, which are also termed balloon cells, are also observed (Robain and Gelot, 1996). HME is probably a heterogeneous condition.
are found throughout the cortex and the underlying white matter. In about 50% of patients, large bizarre cells, which are also termed balloon cells, are also observed (Robain and Gelot, 1996). HME is probably a heterogeneous condition.
HME may be associated with many disorders (see Table 1.3) (Guerrini et al., 1999a), but it can also occur in isolation. The clinical spectrum of HME includes cases with severe epileptic encephalopathy beginning in the neonatal period (Robain et al., 1988) and a few patients who may have a normal cognitive level (Guerrini et al., 1996a; Fusco et al., 1992), with or without epilepsy. The typical presentation is with hemiparesis, hemianopia, mental retardation, and early onset seizures. The most severely affected children have almost continuous seizures beginning in the neonatal period (Fig. 19.4), accompanied or followed by infantile spasms and a burst-suppression
pattern on the sleep EEG (Vigevano et al., 1996; Paladin et al., 1989). A high mortality rate, often due to status epilepticus, is present in the first months or years of life (Robain et al., 1988; Tijam et al., 1978). The survivors have severe cognitive and motor impairment (Trounce et al., 1991). Early hemispherectomy (Di Rocco, 1996) or hemispherotomy (Villemure and Mascott, 1995) might prevent life-threatening seizures or the deleterious interference on the healthy hemisphere (Vigevano et al., 1989a; King et al., 1985). A higher degree of recovery of neuropsychologic function is achieved in patients who are operated on at a young age.
pattern on the sleep EEG (Vigevano et al., 1996; Paladin et al., 1989). A high mortality rate, often due to status epilepticus, is present in the first months or years of life (Robain et al., 1988; Tijam et al., 1978). The survivors have severe cognitive and motor impairment (Trounce et al., 1991). Early hemispherectomy (Di Rocco, 1996) or hemispherotomy (Villemure and Mascott, 1995) might prevent life-threatening seizures or the deleterious interference on the healthy hemisphere (Vigevano et al., 1989a; King et al., 1985). A higher degree of recovery of neuropsychologic function is achieved in patients who are operated on at a young age.
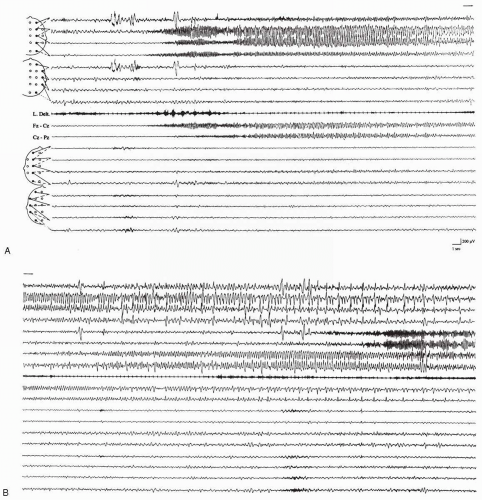 FIG. 19.4. A, B: Seizure activity in hemimegalencephaly. Three-month-old boy (corresponds to Fig. 19.7H) with electrical status epilepticus. Continuous subclinical seizure activity with multiple sites of origin within the malformed hemisphere. |
Focal Cortical Dysplasia
Histologic abnormalities reminiscent of those encountered in HME may be restricted to one cerebral lobe, or they may involve a segment measuring only a few centimeters (Taylor et al., 1971). These discrete cortical malformations were initially described by Taylor et al. (1971) in surgically resected tissue from an epilepsy surgery series. The abnormal area is not usually sharply delineated from the adjacent tissue (Robain, 1996; Taylor et al., 1971). The term focal cortical dysplasia has been used to refer to a wide range of alterations of the cortical mantle. A recent neuropathologic review of a large epilepsy surgery series (Tassi et al., 2002) proposed separating focal cortical dysplasia into the following subtypes: (a) architectural dysplasia, which is characterized by abnormal cortical lamination and ectopic neurones in white matter; (b) cytoarchitectural dysplasia characterized by giant neurofilament-enriched giant neurones in addition to altered cortical lamination; and (c) Taylor-type cortical dysplasia with giant dysmorphic neurones and balloon cells associated with cortical laminar disruption. Such abnormalities probably originate at different times in embryogenesis (Barkovich et al., 2001). In the series by Tassi et al. (2002), patients with architectural dysplasia had a lower seizure frequency than did those with cytoarchitectural and Taylor-type dysplasia.
Focal cortical dysplasia may occur in any part of the cortex (Guerrini, 1997; Guerrini et al., 1992c; Kuzniecky et al., 1992a; Palmini et al., 1991c). The lesions may be quite extensive (see Figs. 3.3 and Fig. 10.3), rendering complete removal impossible in many patients (Olivier et al., 1996; Palmini et al., 1991b). MRI may be unrevealing in up to 34% of patients (Tassi et al., 2002; Desbiens et al., 1993). Distinctive signal alterations on T2-weighted (Fig. 19.5) or FLAIR images are present in most patients with Taylor-type dysplasia, and they are often associated with focal areas of cortical thickening, simplified gyration, blurring of the gray-white limit, or rectilinear boundaries between gray and white matter (Tassi et al., 2002; Kuzniecky, 1996; Bergin et al., 1995). Focal hypoplasia with MRI abnormalities is often found in architectural dysplasia (Tassi et al., 2002). Discrete areas of dysplasia may be associated with widespread minor structural changes of undetermined significance in an undetermined proportion of cases (Sisodiya et al., 1995). Progress in MRI techniques may allow the recognition of subtle areas of dysplasia that are often missed by more conventional studies (Bastos et al., 1999; Sisodiya et al., 1999). Dysplasias involving the mesial temporal lobe are usually accompanied by abnormal development of the hippocampal formation, which sometimes occurs as an isolated anomaly (Baulac et al., 1998).
Neuropathologic and electrophysiologic studies of dysplastic tissue have shown that some dysplasias are intrinsically epileptogenic with continuous or prolonged spike discharges on corticography or depth recordings (Mattia et al., 1995; Palmini et al., 1995). Such discharges might be due to abnormalities of circuitry; these have been demonstrated in some cases (Spreafico et al., 1998; Ferrer et al., 1992).
The clinical presentation in focal cortical dysplasia is typically that of intractable focal epilepsy developing at a variable age but generally before the end of adolescence, although neonatal onset has been reported (Guerrini et al., 1996c; Palmini et al., 1991c). Infantile spasms are common (Dulac et al., 1996; Chugani et al., 1990), but no other age-related epilepsy syndromes are usually observed. Partial status epilepticus has been frequently reported (Palmini et al. 1995; Desbiens et al., 1993; Guerrini et al., 1992c), and, when the location is in the precentral gyrus, the case is often complicated by epilepsia partialis continua (Aicardi, 1994b; Kuzniecky and Powers, 1993; Ferrer et al., 1992; Kuzniecky et al., 1988). Unless the dysplastic area is large, these patients do not suffer from severe neurologic deficits.
The interictal EEG shows focal, often rhythmic epileptiform discharges in about half the patients (Gambardella et al., 1996). These EEG abnormalities (Fig. 19.6) are highly suspicious, they are located over the epileptogenic area, and they are related to the continuous epileptiform discharges recorded with EcoG (Palmini et al., 1995). Most patients with EcoG ictal discharges who have complete removal of the discharging tissue become seizure free or they have a more than 90% reduction in major seizures. None of
the patients with persistence of discharging tissue had a favorable outcome (Palmini et al., 1995).
the patients with persistence of discharging tissue had a favorable outcome (Palmini et al., 1995).
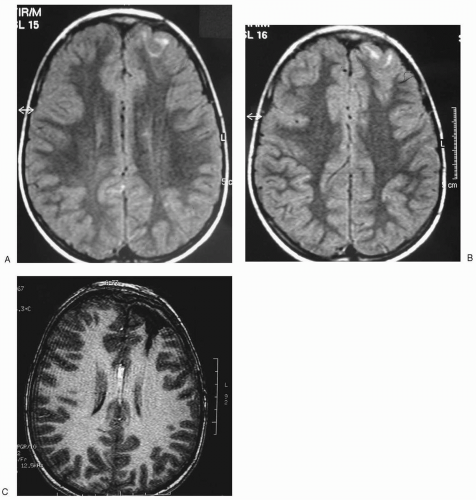 FIG. 19.5. Taylor-type dysplasia (T2) (preoperative and postoperative). Right frontal Taylor-type dysplastic lesion (A, B) in a boy who presented at the age of 3 years with a low number (not more than 10) of episodes characterized by the arrest of ongoing activity and some automatisms. When he was on vigabatrin, he remained seizure free for more than 24 months, and the drug was discontinued. He remained without treatment for 16 months, at which time his epilepsy relapsed. The seizures occurred very frequently. During this same period, he underwent severe behavioral deterioration and regression of school performance. Electroencephalograms (EEGs) showed the previously present right frontal focus, but diffusion of the EEG abnormalities was also present. The previously used and new antiepileptic drugs did not allow control of seizures. After the behavioral deterioration was taken into account, he was operated on 6 months after the relapse (C). He has been seizure free since that time, and he has no behavioral problems. |
The different histologic subtypes of focal cortical dysplasia may carry different chances of seizure freedom after surgery. According to Tassi et al (2002), who used depth electrodes in most cases, patients with Taylor-type dysplasia had the best outcome, with 75% becoming seizure free (Engel class Ia), compared with 50% of those with cytoarchitectural dysplasia and 43% of those with architectural dysplasia. The area of resection is perhaps better defined in patients with Taylor-type dysplasia, possibly due to the distinctive interictal epileptiform discharges (Palmini et al., 1995), which can be captured by depth electrodes (Munari et al., 1996).
However, the clinical and electrophysiologic features reported in focal cortical dysplasia and HME are biased because they originate from epilepsy surgery centers at which the most severe cases are seen. In the authors’ experience, some patients with
well-controlled seizures show focal dysplastic lesions on MRI that are identical to those present in patients with histologically proven focal dysplasia.
well-controlled seizures show focal dysplastic lesions on MRI that are identical to those present in patients with histologically proven focal dysplasia.
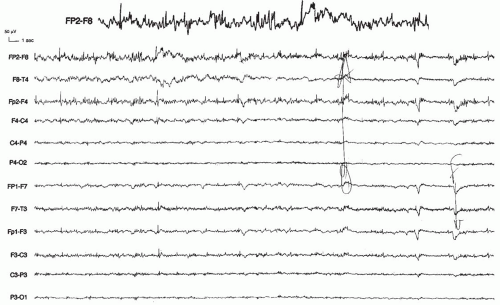 FIG. 19.6. Interictal electroencephalographic abnormalities (Fp2-F8) related to the presence of a right frontal cortical dysplasia. (Courtesy of Dr. Pierre Thomas, Nice University Hospital, France.) |
Schizencephaly
Schizencephaly (cleft brain) consists of a full-thickness unilateral or bilateral cleft of the cerebral hemispheres with communication between the ventricle and the pericerebral subarachnoid spaces. The cortex surrounding the cleft is comprised of polymicrogyria, which also covers the lips of the fissure and extends along the walls of the cleft all the way to the ventricular surface (Ferrer, 1984) and which is probably responsible for the epilepsy. The walls of the clefts may be widely separated (open-lip schizencephaly) (Fig. 19.7A) or closely apposed (closed-lip schizencephaly) (Fig. 19.7B). The clefts may be located in any region of the hemispheres, but, by far, they are most commonly located in the perisylvian
area (Barkovich and Kjos, 1992). The bilateral clefts are usually symmetric in location but not necessarily in size. Unilateral clefts are sometimes associated with a localized cortical abnormality in the contralateral hemisphere. Septooptic dysplasia (agenesis of the septum pellucidum and optic nerve hypoplasia) is observed in some patients (Barkovich and Norman, 1988). No agreement exists on whether schizencephaly should be classified as an abnormality of neuron proliferation (Barkovich et al., 2001), and establishment of the time of origin of the developmental abnormality during embryonic development is extremely difficult.
area (Barkovich and Kjos, 1992). The bilateral clefts are usually symmetric in location but not necessarily in size. Unilateral clefts are sometimes associated with a localized cortical abnormality in the contralateral hemisphere. Septooptic dysplasia (agenesis of the septum pellucidum and optic nerve hypoplasia) is observed in some patients (Barkovich and Norman, 1988). No agreement exists on whether schizencephaly should be classified as an abnormality of neuron proliferation (Barkovich et al., 2001), and establishment of the time of origin of the developmental abnormality during embryonic development is extremely difficult.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


