Fig. 1
Post SAH patient mortality and underlying mechanisms: mortality in SAH patients is greatest in the early phase (a). Although early mortality results from early activating mechanisms, delayed mortality is the product of both the early and the delayed developing complications (b). See text for explanation
Two phases of injury after SAH are recognized: the early phase, which begins at aneurysmal rupture and extends through the first 3 days, and the delayed phase, which appears 3–14 days later and is characterized by delayed vasospasm and delayed neurological deficits. Studies show that early injury is the product of the events that occur at or immediately after SAH. These key events can broadly be categorized as: blood release, acute reactive hyperemia, and transient global ischemia (Fig. 2). Each of these event triggers mechanisms that are specific to it or are shared among the events to create early brain injury (Fig. 3). These mechanisms may evolve with time and contribute to delayed phase complications. Here, we briefly review key events after SAH and the mechanisms that they activate.
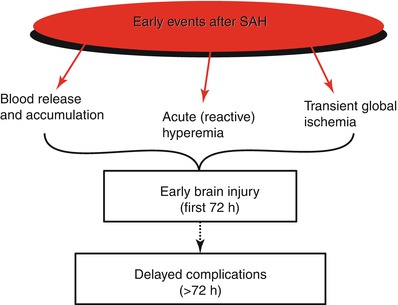
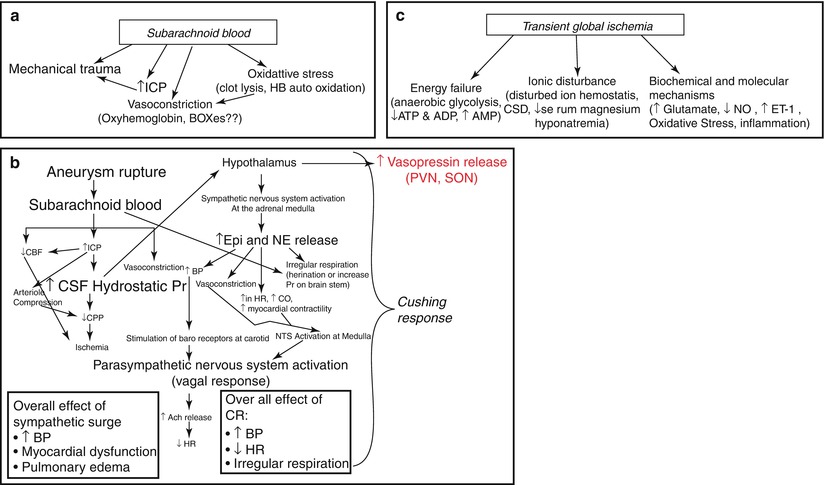
Fig. 2
The early events after SAH: broadly, early events after SAH can be divided as: release of blood, acute reactive hyperemia representing the Cushing response, and transient global ischemia. Each of these events triggers mechanisms that contribute to early brain injury and evolve with time to participate in delayed complications after SAH
Fig. 3
Mechanisms of early brain injury after SAH: (a) outlines the mechanism by which subarachnoid blood participates in early brain injury after SAH; (b) outlines the triggers of the Cushing response (CR) after SAH, mechanisms that the CR activates, and the complications that follow; and (c) outlines the main mechanism by which transient global ischemia contributes to early brain injury after SAH
Subarachnoid Blood
The volume of subarachnoid blood correlates with the patient’s status at admission and with neurological deficits and clinical outcome [2]. Blood can damage the brain by inducing mechanical and chemical trauma (Fig. 3a).
Mechanical Trauma
Mechanical trauma occurs as blood that is released under high pressure stretches the subarachnoid space, compresses the surrounding arteries, and elevates the intracranial pressure. Cerebral vasculature is very sensitive to changes in local environment and vessels constrict as they come in contact with blood. A short-lived constriction of large cerebral arteries is frequently observed in the first minutes after experimental SAH [3]. Blood, in addition, obstructs the passage of cerebrospinal fluid (CSF) and hence increases the risk of acute hydrocephalus, which is observed in as many as 50 % of SAH patients and is associated with poor outcome [4].
Chemical Trauma
Chemical trauma is created by the products of subarachnoid blood lysis. Blood lysis begins within minutes after SAH and results in the appearance of oxyhemoglobin and xanthochromia in the CSF. Hence, xanthochromia is often used for clinical SAH diagnosis when CT scan results are negative [5]. At least three major products of clot lysis are implicated in SAH pathology: oxyhemoglobin, iron, and the recently discovered bilirubin oxidation products (BOXes).
Oxyhemoglobin
Oxyhemoglobin levels peak in the CSF at 24 h after SAH and decrease thereafter (from animal and patient cerebral microdialysis studies). Oxyhemoglobin is a potent spasmogen and is implicated in early arterial constriction and in delayed vasospasm after SAH [2, 6]. Studies that report that in vivo (subarachnoid space) and in vitro (basilar artery) exposure to oxyhemoglobin creates ultrastructural alterations in vascular morphology similar to those documented early after SAH support this implication [7]. These alterations include the appearance of cell craters, blebs, and vacuoles in endothelial cells and detachment of endothelium from the basement membrane [8]. Oxyhemoglobin is also cytotoxic to endothelial cells and astrocytes [9, 10]. The mechanisms underlying oxyhemoglobin-induced constriction include scavenging and creating a deficiency of nitric oxide (endothelial and neuronal) [11], suppression of voltage-dependent potassium currents [12], and an increase in endothelin (ET)-1 production [13].
Heme Metabolism Products
Heme is metabolized by heme oxygenase (HO)-1 into iron, biliverdin, and carbon monoxide [14].
Iron
Approximately 70 % (1,800 mg) of the body’s iron is bound to hemoglobin in the form of heme; each hemoglobin molecule has four heme groups. As a result, a substantial amount of iron is released into the subarachnoid space after lysis of subarachnoid blood. HO-1 (see above) is upregulated and the expression of ferritin, an iron-binding protein, increases 1–3 days after SAH [15, 16]. The ferritin level in the CSF is considered a good diagnostic measure for SAH patients who present late to the clinic [17]. After SAH, ferritin expression increases in microglia, neurons, and cells in the adventitia of the arterial wall [15, 16, 18]. Lee et al. used an iron chelator, deferoxamine, to establish a role of iron in oxidative injury after SAH [18]. In this experiment, deferoxamine administered 3–6 h after SAH reduced cerebral iron deposits, HO-1 and ferritin expression, oxidative stress and neuronal apoptosis, and extended animal survival [18].
Bilirubin Oxidation Products (BOXes)
BOXes, as the name indicates, are formed upon oxidative degradation of bilirubin [19]. Bilirubin is formed by the action of biliverdin reductase on biliverdin (a product of heme metabolism; see above). BOXes are potent vasoconstrictors and are present in significantly higher concentration in the CSF of SAH patients who develop vasospasm than in the CSF of the patients who do not [20]. Hence, BOXes are being studied for their possible contribution to the pathogenesis of delayed vasospasm. A role of BOXes in early brain injury is anticipated but remains to be elucidated.
Acute Reactive Hyperemia; Cushing Response (CR)
The Cushing response (CR) is a compensatory hypothalamus-mediated response to a rise in intracranial pressure (ICP). The severity of the CR at SAH depends on the magnitude of rise in ICP, which in turn depends upon the volume of subarachnoid blood. The CR acts to maintain the difference between blood pressure (BP) and ICP to oppose drops in cerebral blood flow (cerebral perfusion pressure (CPP) = BP − ICP); however, it is not always successful [21]. In SAH patients, the CR is mostly observed as a sympathetic surge with increased serum epinephrine (EP) and/or norepinephrine (NE) levels. Two major complications, myocardial dysfunction and pulmonary edema, are associated with sympathetic surge [22] (Fig. 3b). These complications develop early after SAH and are often associated with poor patient outcome.
Cardiac Complications
In most SAH patients, cardiac complications appear immediately or develop within a few hours after aneurysmal rupture [22]. These complications include electrocardiographic (ECG) abnormalities, cardiac arrhythmias, and myocardial damage. ECG abnormalities develop within the first 48 h after SAH and reappear at rebleed [23–25]. Prominent ECG abnormalities include P wave abnormalities, prolonged QT interval, ST segment, and T-wave and are associated with poor neurologic grade on admission [26]. Cardiac arrhythmias can occur as the first symptom of acute SAH and are life threatening in 5–10 % of cases [27]. Major rhythmic abnormalities include sinus tachycardia, sinus bradycardia, and premature atrial and ventricular beats [25]. Myocardial injury is found in approximately 30–60 % of SAH patients and is associated with higher mortality [28–30]. Serum creatine kinase (CK)-MB and troponin-1 concentrations that are associated with myocardial injury are frequently increased within 24–48 h in SAH patients [28–30]. In addition, contraction band necrosis, myocardial fragmentation, and focal myocytolysis in the myocardium is found in autopsy of patients who died early after SAH [29–32]. In animals, increased serum CK-MB and cardiac troponin-1 are found as early as 10 min [33], and myocardial damage at 3 h, after SAH [32].
Pulmonary Complications
Pulmonary edema, also called neurogenic pulmonary edema, occurs in 10 % of all SAH cases and is a predominant complication in poor grade (Hunt and Hess grade III–V) patients [34]. Pulmonary edema usually develops within hours after SAH; however, a delayed form that appears 3–5 days after SAH is also reported [35]. In animals, leakage of lung capillaries occurs 8 min after injection of blood into the cisterna magna [36]. The nature of mechanisms underlying SAH-related pulmonary edema are cardiogenic (myocardial dysfunction leading to a raised hydrostatic pressure and fluid retention), noncardiogenic (such as acute lung injury or acute respiratory distress syndrome), and neurogenic (pressure of blood on respiratory centers at SAH) [34, 35].
Transient Global Ischemia (TGI)
Ischemia after SAH affects the entire brain and appears early. Mechanisms triggered by SAH-induced transient global ischemia (TGI) can be broadly classified as energy failure, ionic disturbances, and biochemical and molecular mechanisms (Fig. 3c). A brief description of these mechanisms is given below; for a review that is more detailed, an interested reader is referred to recent reviews [37, 38].
Energy Failure
Energy failure after SAH is mostly observed as changes in cerebral energy metabolism as detected in cerebral microdialysis studies. In addition, depletion of high-energy phosphate stores (fall in ATP and ADP and rise in AMP) in the cerebral cortex is noted as early as 6 h after experimental SAH [39, 40]. Cerebral microdialysis studies in animals have established that energy metabolism is disturbed within minutes after SAH, causing depletion of pyruvate and glucose and rises in lactate and in the lactate-to-pyruvate ratio [41, 42]. Similar findings are made in clinical SAH. Sarrafzadeh and colleagues noted that the lactate-to-pyruvate ratio remains significantly high for the first 8 days after SAH in patients with acute ischemic neurological deficits [42].
Ionic Changes
Energy is required to fuel ATPases to maintain ionic homeostasis across cell membranes. After SAH, as brain energy stores deplete, rapid alterations in ionic distributions across the membranes of brain cells and of cerebral vascular cells occur. In metabolically active neurons and astrocytes, as Na+K+-ATPases cease to work, intracellular Na+ rises and, on reaching a critical point, reverses the operation of the Na+–Ca2+ exchange carrier, creating increases in intracellular Ca2+ and extracellular K+. Changes in neuronal ionic content promotes the cortical spreading depolarization and disturbed neurovascular coupling that contributes to the acute pathophysiology of SAH and the later-occurring delayed ischemic neurological deficits (DINDs) [43]. In cerebral endothelial and smooth muscle cells, a pathological rise in intracellular calcium promotes constriction [44, 45]. Other pathological consequences of rises in intracellular calcium include the release of neurotransmitters such as glutamate and activation of enzymes, including ones that are detrimental to cells, such as inducible nitric oxide (NO) synthase (iNOS) and caspases [46, 47].
Biochemical and Molecular Mechanisms
Although the list of biochemical and molecular mechanisms that activate early and contribute to SAH pathology is long, the key mechanisms are as follows:
Extracellular Glutamate
Animal cerebral microdialysis studies show that interstitial glutamate concentration increases after SAH, reaches a peak at 40 min, and is associated with early pathology [48, 49]. Clinically, microdialysis studies have found that, during the first 8 days after SAH, the cerebral glutamate concentration correlates with early and delayed status of SAH patients; the concentration is greatest in the patients who develop acute ischemic neurological deficits, and is elevated (although not as much) in patients who develop delayed ischemic neuronal deficits [42].
Nitric Oxide (NO)
Cerebral NO levels are altered after SAH. Cerebral NO levels decrease in animals minutes after SAH and increase at 24 h [11, 50]. Sehba et al. found that an NO donor administered early after SAH recovers the cerebral blood flow (CBF) reduction and prevents ischemic glutamate release in animals after SAH [49]. In the clinic, the initial increase in cerebral NO has not been studied, but an increase in NO level in the CSF at 24 h after SAH has been established [51, 52]. Patients with low NO levels in the CSF at 0–12 days after SAH are more prone to develop delayed vasospasm [51, 52].
Endothelin (ET)-1
An increase in CSF ET-1 is observed within minutes in animals and within 24 h in patients after SAH [53]. Because ET-1 is a potent spasmogen that produces long-lasting constriction, the potential of receptor antagonists in preventing the pathogenesis of delayed vasospasm after SAH was evaluated. These clinical trials successfully reduced/inhibited development of the delayed vasospasm but did not improve patient’s outcomes [54, 55].
Inflammation
The status of acute mediators of inflammation, cells of the immune system (such as neutrophils and platelets), pro-inflammatory cytokines (interleukin (IL)-1β, IL-6, tumor necrosis factor (TNF)-α, and others), and adhesion molecules (intercellular adhesion molecule (ICAM)-1, P and S-selectins, and others), is studied after SAH. Animal studies establish overt vascular neutrophil accumulation, serum cytokine concentration, and expression of adhesion molecules within the hours after SAH and their associations with early brain injury [37]. In patients, similar increases in serum and CSF cytokines and in soluble forms of adhesion molecules (within 1–3 days from ictus) are established and associated with hyperthermia, vascular spasm, and unfavorable outcome [56–58].
Oxidative Stress
Oxidative stress after SAH is observed as the presence of lipid peroxidation products, decreased activities of enzymatic and non-enzymatic antioxidant systems, and generation of reactive oxidative species. In experimental animals, oxidative stress is present within the first 24 h [59] and, in patients, within the first 72 h after SAH [60]. In patients, an increase in lipid peroxidation products during the early phase of SAH is associated with the pathogenesis of delayed vasospasm and with poor outcome [61, 62].
Conclusion
In conclusion, a number of key events are activated within the first 72 h after SAH and trigger mechanisms that contribute to early brain injury and are also associated with delayed vasospasm and overall outcomes of patients. A better understanding of early mechanisms and their timely prevention may provide a superior strategy to improve clinical outcome in this patient population.
Related posts:
 Value of Perfusion Computed Tomography (PCT) Imaging After Aneurysmal Subarachnoid Hemorrhage: A Review of the Current Data
of Patients Without Neuropsychological Deficits Following Aneurysmal Subarachnoid Haemorrhage
Combined Endovascular Therapy with Milrinone and Nimodipine for the Treatment of Severe Vasospasm: Preliminary Results
Rate in the Sylvian Cistern Is Associated with the Severity of Cerebral Vasospasm After Subarachnoid Hemorrhage
of Human Albumin on TCD Vasospasm, DCI, and Cerebral Infarction in Subarachnoid Hemorrhage: The ALISAH Study
Cisterna Magna Double-Injection Model of Subarachnoid Hemorrhage – Background, Advantages/Limitations, Technical Considerations, Modifications, and Outcome Measures
Value of Perfusion Computed Tomography (PCT) Imaging After Aneurysmal Subarachnoid Hemorrhage: A Review of the Current Data
of Patients Without Neuropsychological Deficits Following Aneurysmal Subarachnoid Haemorrhage
Combined Endovascular Therapy with Milrinone and Nimodipine for the Treatment of Severe Vasospasm: Preliminary Results
Rate in the Sylvian Cistern Is Associated with the Severity of Cerebral Vasospasm After Subarachnoid Hemorrhage
of Human Albumin on TCD Vasospasm, DCI, and Cerebral Infarction in Subarachnoid Hemorrhage: The ALISAH Study
Cisterna Magna Double-Injection Model of Subarachnoid Hemorrhage – Background, Advantages/Limitations, Technical Considerations, Modifications, and Outcome Measures
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


