Fig. 34.1
Oneiric stupor. Sequential frames recorded from top to bottom in patients with fatal familial insomnia ( FFI), Morvan syndrome ( MS) and delirium tremens ( DT) showing similar complex semi-purposeful movements during oneiric stupor episodes
From the early stages of disease, patients also present profuse sweating, abundant salivation and lacrimation and a progressive increase in heart and breathing rates accompanied by a mild but progressive rise in body temperature. Associated disorders include difficulty urinating and impotence in males [1, 12].
As the weeks pass, difficulty standing, a complex gait impairment, distal tremors and tendon hyperreflexia become increasingly evident. Worsening somnolence is transformed into a permanent state of drowsiness (“dormiveglia”) or stupor persisting night and day. When stimulated, patients open their eyes and answer correctly but in monosyllables. Death is due to sudden cardiorespiratory arrest or intercurrent febrile illness.
The disease has a rapid course (8–12 months) in around two-thirds of patients. In these cases, both mutated and non-mutated alleles of PRNP at the position 178, and at the polymorphic codon 129 encode methionine [13]. In the remaining cases in which the non-mutated allele in position 129 of the PRNP encodes valine, the clinical course is at least two to three times longer and highly variable.
Serial neuropsychological tests have documented that attention and vigilance progressively decline from the early signs of disease, whereas intellectual functions remain largely spared until the most advanced stages. Given these characteristics, FFI should be considered a chronic confusional state rather than a form of dementia [14, 15].
Serial electroencephalogram (EEG) performed throughout the disease course fail to disclose abnormalities other than those linked to a progressive increase in somnolence. Serial polygraphic recordings for 24 h during disease evolution document a fragmentation and progressive reduction of spindle and delta activities that ultimately disappear completely [16]. Once spindle and delta activities disappear, intravenous injection of benzodiazepines or barbiturates at high doses transform “dormiveglia” or stupor into a deep coma associated with flattening of the EEG, invariably failing to trigger spindle and delta activities [17].
This state of subvigilance persisting day and night is characterized polygraphically by an alternation of stage 1 sleep (EEG theta activity associated with slow eye movement discharges and diminished muscle tone) and very short bursts of rapid eye movement (REM) sleep (lasting 30–40 s) that recur, often in clusters, both day and night. Stage 1 and REM sleep continue to alternate until the preterminal stages of the disease (Fig. 34.2).
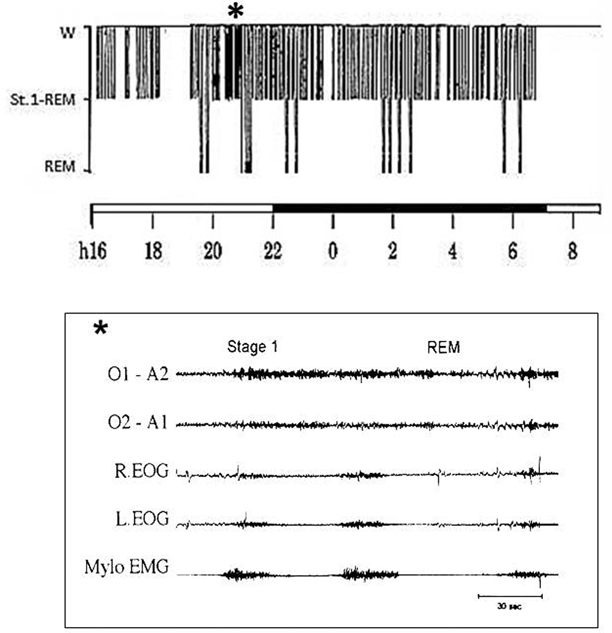
Fig. 34.2
Polysomnographic recording in fatal familial insomnia. Top: The 24-h wake–sleep histogram demonstrates reduced total sleep time, absence of synchronized sleep and abnormal cyclic organization of sleep. Bottom: Electroencephalographic tracing shows transitions between rapid eye movement ( REM) and subwakefulness (stage 1) recurring in a quasi-periodic fashion
Background and stimulated cardiovascular function monitoring documents a state of sympathetic hyperactivity whereas parasympathetic activity is always within normal limits [18]. Serial 24-h recordings disclose a progressive increase in heart and breathing rates, systemic arterial pressure and body core temperature during the clinical course [19]. Circadian cortisol and noradrenaline secretions are two to three time higher than normal, daily and nightly, whereas the nocturnal melatonin peak subsides to almost disappear [20] (Fig. 34.3).
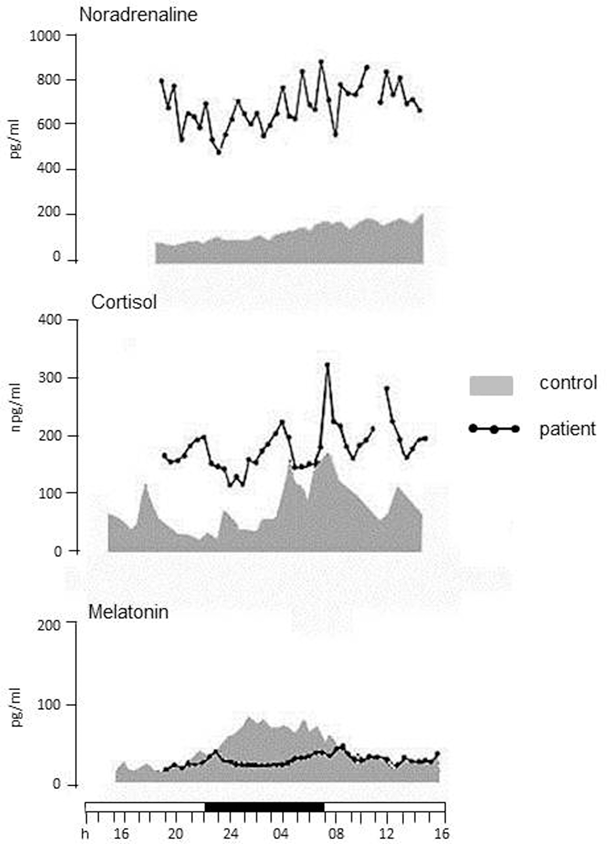
Fig. 34.3
The 24-h recordings of noradrenaline, cortisol and melatonin concentrations in a patient with FFI and in a control individual. Noradrenaline and cortisol secretions are higher than normal throughout the 24 h. The nocturnal peak of melatonin secretion is lost
Computed tomography (CT) and magnetic resonance imaging (MRI) scans are unremarkable except for a mild cerebral and cerebellar atrophy in the most advanced disease stages. By contrast, serial positron emission tomography (PET) (18fludeoxyglucose(FDG) PET) scans invariably show a bilateral thalamic hypometabolism from the early disease stages [21, 22]. Impaired thalamic metabolism is evident many months before the onset of the early signs of disease in carriers of the FFI mutation [23].
General autopsy fails to disclose findings of significance whereas brain examination reveals bilateral and symmetrical atrophy of the thalami. The thalamic formations invariably and most severely affected are the anterior and medio-dorsal thalamic nuclei. Neuronal loss in these nuclei exceeds 50 %, reaching values of more than 90 % in the antero-ventral nuclei and mesial part of medio-dorsal nuclei. The other nuclei, with the exception of the pulvinar, invariably damaged, are inconsistently and scarcely affected.
According to Schulman [24], who observed an ante-litteram FFI case, and Macchi et al. [25] who studied two FFI patients by morphometric analysis, the thalamic reticular nucleus (TRN) was one of the most damaged thalamic formations. However, Gambetti (personal communication) claimed it would be difficult if not impossible to establish whether the entire length of this extremely long and thin layer is involved in the pathological process.
Rapid evolution cases often present areas of spongiform degeneration confined to the anterior cingulate gyrus and mesio-orbital cortical and sub-cortical formations [26]. In longer evolution cases, cortical spongiform degeneration may extend throughout the frontal lobe to the parieto-temporal regions, but always prevails in the mesio-orbital areas of the frontal lobe. FFI can be defined as a thalamo-limbic or prevalently thalamo-limbic encephalopathy .
Genetic and Molecular Biology of FFI and Other Prion Diseases
As mentioned above, FFI is a prion disease caused by a point mutation at codon 178 of PRNP. The FFI phenotype is seen when the methionine–valine polymorphic codon 129 of the PRNP encodes methionine in the mutated allele. If the mutated allele at codon 178 encodes valine, at position 129, the disease phenotype will be a rapidly evolving dementia associated with diffuse spongiform degeneration of the cortex and subcortical grey matter structures [11].
The most common FFI phenotype (short course and anatomic lesions confined to the thalamus) presents when codon 129 of the prion protein gene encodes methionine in mutated and non-mutated alleles (around two-thirds of patients). When the non-mutated allele encodes valine (methionine–valine heterozygous at codon 129), the disease has a longer more variable course with thalamic atrophy associated with spongiform degeneration of the frontal and parieto-temporal cortices. The prion protein fragment deposited in the brain tissues of FFI patients has a molecular weight and conformation different from that of the fragments found in the brains of patients with the spongiform encephalopathy variant caused by the same mutation at codon 178, co-segregating with valine in the mutated allele. For the first time in human genetics, these molecular and genetic findings showed that the disease generated by a punctiform mutation of the prion protein gene is due to a polymorphism of the same gene [13]. Working with Prusiner and his group, we were able also to demonstrate that transgenic mice infected with FFI brain homogenates develop and transmit a disease characterized by prions that retain the donor prion conformation. Conversely, transgenic mice infected with brain homogenates of patients with Creutzfeldt–Jakob disease develop and transmit a diffuse encephalopathy characterized by prions with a conformation similar to that of the donor [27]. These findings provide convincing evidence of Prusiner’s theory that the variable clinical manifestations of prion disease are linked, at least in part, to different prion conformations rather than a hypothetical prion-driven virus.
Clinical and Polygraphic Aspects of Agrypnia Excitata
Some 15 years after the first description of FFI, we realized that at least two other neurological disorders, MS and DT , present features similar to FFI consisting of a confusional state characterized by a polygraphic tracing with intermediate aspects between stage 1 NREM sleep and REM sleep. Stage 1 + REM or stage 1 REM were the terms used by Tachibana et al. and Kotorii et al. to define the polygraphic tracing they observed in cases of acute psychosis triggered by untreated sudden alcohol withdrawal in patients with chronic alcoholism [28, 29].
In addition, FFI, MS and DT share the features of a syndrome characterized by a general and permanent state of motor and autonomic activation. We named this condition AE . The hallmark of AE is the stereotyped subcontinuous repetition of gestures mimicking the daily-life activities described above under the name of OS (Fig. 34.1).
OS, a sign we invariably observed in cases of FFI, MS and DT, but never encountered in patients with confusional states of different nature and origin, is the most characteristic feature of AE.
Pathophysiology of AE
In FFI, AE is caused by a breakage in the circuit running through the medio-dorsal thalamic nuclei and rostral part of the TRN and interconnecting the basal forebrain formation to hypothalamus and upper brainstem. In MS, autoantibodies binding to subunits of VGK channels, particularly expressed by GABAergic neurons of the thalamus, impair normal thalamic function [8, 30, 31]. This mechanism is responsible for the loss of sleep associated with day and night motor , sympathetic and noradrenergic activation in MS [8, 30, 31]. In DT, the disappearance of sleep combined with a general hyperactivation syndrome ensues from a concomitant downregulation of GABA-A synapses and upregulation of N-methyl-d-aspartate (NMDA) glutamatergic synapses following untreated sudden alcohol withdrawal in chronic alcohol abusers [9].
Related posts:
 Sleep in the Biblical Period
Sleep Medicine Around the World (Beyond North American and European Continents, and Japan)
Sleep in the Biblical Period
Sleep Medicine Around the World (Beyond North American and European Continents, and Japan)
 The Greco-Roman Period
Sleep Deprivation: Societal Impact and Long-Term Consequences
Psychological Treatment of Insomnia: The Evolution of Behavior Therapy and Cognitive Behavior Therapy
The Greco-Roman Period
Sleep Deprivation: Societal Impact and Long-Term Consequences
Psychological Treatment of Insomnia: The Evolution of Behavior Therapy and Cognitive Behavior Therapy
 Women’s Health and Sleep Disorders
Women’s Health and Sleep Disorders
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


