Nosological classification of organic dementia is based on current knowledge and theories of aetiology, including genetics, clinical picture, the pathological substrate, and the predominant location of brain damage. This chapter is concerned with dementia syndromes caused by a degenerative disease primarily affecting the frontal and temporal lobes, named frontal-lobe dementia(1) or frontotemporal dementia (FTD).(2) The terminology should be viewed from a historical perspective. The relationship between localized cortical atrophy in dementia and symptoms of aphasia was first reported by Pick in 1892.(3) The pathological account of this lobar degeneration by Alzheimer in 1911 described ‘ballooned’ neurones (Pick cells) and argentophilic globes (Pick bodies),(4) and the clinicopathological entity was named Pick’s disease.
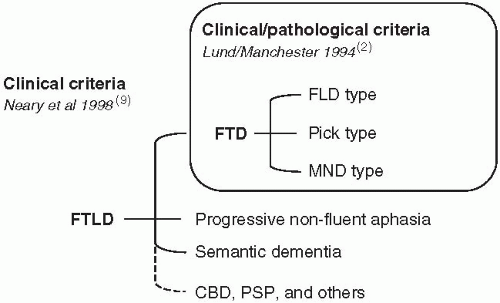
In the 1980s, attention was drawn to a larger group of frontal-lobe dementias associated with frontotemporal cortical degeneration.(5,6,7) The Lund-Manchester consensus of 1994 delineated the prototypical clinical syndrome of FTD with three neuropathological constituents, frontal lobe degeneration of non-Alzheimer type (FLD),(5) (alternatively designated ‘dementia lacking distinctive histology’),(8) Pick’s disease, and motor neurone disease (MND) with dementia (FTD-MND).(2) The 1998 consensus on clinical diagnostic criteria for frontotemporal lobar degeneration (FTLD)(9) encompassed two additional dementia syndromes; progressive non-fluent aphasia (PA),(10,11) and semantic dementia.(12) Corticobasal degeneration (CBD) and progressive supranuclear palsy (PSP) have also been associated with FTLD.(13) A changing clinical classification is shown in Fig. 4.1.3.1. The addition of important genetic and histochemical characteristics has further added to the complex classification of FTD and FTLD with a risk of developing numerous and partly competing definitions. FTLD may be further subclassified into forms positive or negative for tau and ubiquitin. The ubiquitinated form will be referred to as FTD-U, which is synonymous to FLTD-U.(14)
Neuropathology
On a neuropathological basis about two-thirds of the FTLD cases are of the type with ubiquitinated inclusions (FTD-U) or of FTD type without such inclusions (FLD), both lacking tau pathology.(14,15) In an attempt to classify FTD forms from a structural point of view, FLD might be appointed as a basic form, showing type and distribution of main pathological changes common to the majority of FTLD forms. To this set of alterations are added further features proceeding in the description of frontotemporal degenerative disorders (Table 4.1.3.1).
In FLD, the cortex is the site of a simple degenerative process resulting in a cortical atrophy which is frontotemporal with or without asymmetries, moderate or even at times mild. It involves cortical layers 1-3, showing neuronal loss, gliosis, and microvacuolation, seen also in the striatum in a small proportion of cases as well as a mild-to-moderate degeneration of the substantia nigra.(5) DLDH has essentially the same pathology as FLD.(8)
Fig. 4.1.3.1 Clinical classification of frontotemporal dementias.
In FTD-U the ubiquitin-positive inclusions and dystrophic neurites supplement the picture of FLD, as also described in FTD, linked to chromosome 3.(16) The inclusions contain a ubiquitinated protein, also identified in some other varieties of FTD such as FTD-MND.(17) Here there is also a degeneration of motor neurones and paths. The language variants of FTLD show a mainly temporal degeneration in semantic dementia and an asymmetric frontotemporal degeneration with left-sided predominance in PA.
With a mainly frontal or frontotemporal including anterior cingulate gyrus involvement FTD differs markedly from Alzheimer’s disease (AD) with its posterior temporal parietal, posterior cingulate, and severe hippocampal involvement and also with Lewy bodies, amyloid, and plaque pathology not seen in FTD. So far mentioned FTD forms belong in the same histopathological group of non-tauopathies and further share a frequent and individually varyingly severe white-matter sclerosis, which is often predominantly frontal and may be primary or secondary.(18) It is also seen in the tauopathy group and in forms of vascular dementia.
The remaining, less common forms comprise the FTD-tau group. In Pick’s disease the degenerative process is more intense and partly involves all cortical layers, creating a severe circumscribed or lobar frontotemporal atrophy referred to as ‘knife blade atrophy’. Microscopically there are tau-positive neuronal inclusions and sometimes glial tangles and often ballooned nerve cells. The FTD with Parkinsonism (FTDP-17) group with a number of familial disorders shares basic pathological features with the FLD but is regularly more severe in the basal ganglia and substantia nigra.(19) Glial cells and neurones contain various types of tau-positive inclusions. CBD is structurally and clinically heterogeneous, resulting in overlap with other diseases, especially PSP.(20) These two may represent varieties of the same pathological process, less obviously belonging in the FTD group even if a similar laminar frontal cortical degeneration and related symptoms are part of the presentation. In CBD ballooned neurones, tau-positive inclusions in neuronal and glial cells, white-matter rarefaction and nigral degeneration may be found. For PSP different forms and patterns are noted but it basically affects more widely the striatum, basal ganglia, and hypothalamus. It also involves the brain stem including the substantia nigra as well as the cerebellum, again with silver- or tau-positive inclusions and tangles in neurones and glial cells.(21)
Table 4.1.3.1 Organic dementia with frontotemporal lobar degeneration (FTLD)
Frontal lobe degeneration of non-Alzheimer type (FLD) (Dementia lacking distinctive histology) without ubiquitinated inclusions
FTD with ubiquitin-positive inclusions (FTD-U)
Familial FTD with chromosome 3 mutation (FTD-3)
FTD with motor neurone disease (FTD-MND)
Progressive non-fluent aphasia (PA)
Semantic dementia
Pick’s disease
FTD with Parkinsonism (FTDP-17)
Corticobasal degeneration (CBD)
Progressive supranuclear palsy (PSP)
Epidemiology
Most demographic data concern the grouping of FTD, not separating FLD and Pick’s disease. Pick’s disease is rare, estimated at 24-60/100 000 in Minnesota and 30-60 in Switzerland.(22) The calculated prevalence of FTD in the Netherlands is 10.7 per million between 50 and 60 years of age and 28 between 60 and 70 years.(23) The prevalence of FTD in the province Zuid-Holland in the Netherlands was 3.6/100 000 at age 50-59, 9.4 at age 60-69 years, and 3.8 at age 70-79 years.(24) Pasquier et al.(25) diagnosed FTD in 4.8 per cent of all types of dementia. The marked geographic variation of the prevalence might be due to genetic and environmental factors, but also influenced by differences in the diagnostic process and the age group studied. In a clinical study of a total catchments area of 20 million people in Germany the relative proportion of FTLD was 1.9 per cent.(26) The prevalence of dementia in motor neurone disease has been estimated to 2-6 per cent.(27) The proportion of FTD in relation to all types of dementia in different clinicopathological studies varies between 5 and 18.9 per cent.(7,8,28,29,30)
Clinical features
The first clinical manifestations of FTD usually appear in the presenium, in some cases as early as 35 and seldom after 70 years of age. The mean age at onset in post-mortem verified FLD cases is 56 ± 7.6 years with a mean duration of 8 ± 3.4 years (range 3-17 years).(31) The mean age of onset in Pick’s disease is similar, 62 years, with a range of 40-80 years and a mean survival of 9.8 years with a range of 4.8-21.2 years.(22) The large variations of the duration of FLD and Pick’s disease are similar to that of earlyonset AD. The clinical onset of MND dementia is usually in the sixth decade and the mean duration is about 30 months. Age at onset is similar in familial and sporadic cases of FTD and sometimes past 80 years.(32) The Lund-Manchester consensus on clinical criteria for FTD is summarized in Table 4.1.3.2.
Disordered behaviour
The early stage of FLD and Pick’s disease is characterized by changes of personality and behaviour, affective symptoms, and a progressive reduction of expressive speech. The clinical onset is insidious with slow progression without ictal events. The changes of personality and behaviour are rather non-specific and easily misinterpreted as a non-organic mental disease such as mood disorder, stress reaction, schizophrenia, or other psychotic reaction. Loss of insight concerning the mental changes is an early and alarming manifestation of the disease. FTD patients may, however, consult a doctor referring to symptoms such as anxiety, tiredness, and strange somatic complaints combined with bizarre hypochondriacal ideas.
Table 4.1.3.2 The Lund-Manchester consensus (1994) on clinical criteria for frontotemporal dementia (slightly modified)(2)
Core diagnostic features
Behavioural disorder
Insidious onset and slow progression
Early loss of insight into changes of own mental state
Early loss of personal and social awareness
Early signs of disinhibition and lack of judgement
Mental rigidity and inflexibility
Stereotyped, repetitive, and imitating behaviour
Hyperorality, oral/dietary changes
Utilization behaviour
Distractibility, impulsivity, and impersistence
Affective symptoms
Depression, anxiety, excessive sentimentality
Hypochondriasis, bizarre somatic complaints
Emotional bluntness, apathy, and lack of empathy
Amimia
Speech disorder
Progressive reduction of speech output
Stereotypy of speech, perseveration
Echolalia
Late mutism
Spatial orientation, receptive speech, and praxis preserved
Neuropsychology: profound failure on ‘frontal-lobe’ tests in the absence of severe amnesia, or perceptual spatial disorder
Supportive diagnostic features
Onset before 65
Positive family history of similar disorder in a first-degree relative
Bulbar palsy, muscular weakness and wasting, fasciculations (motor neurone disease)
(Reproduced from A. Brun, E. Englund, L. Gustafson, et al. Consensus statement— clinical and neuropathological criteria for frontotemporal dementia, Journal of Neurology, Neurosurgery, and Psychiatry, 57, 416-18, copyright 1994, BMJ Publishing Group Ltd.)
The early loss of personal and social awareness is seen as neglect of personal hygiene and grooming, and tactlessness and antisocial behaviour.(6,9) The impaired control of behaviour is seen as increased sentimentality, inadequate smiling, inappropriate joking, irritability, and acts of aggressiveness, leading to conflicts at home and work. Craving for affection and sexual contact may be easily provoked, but usually expressions of sexual disinhibition are possible to divert. Impulse buying, shoplifting, indecency, and other disinhibited behaviour may, however, lead to rejection by the family and society. Such unpredictable and pseudopsychopathic behaviour imposes severe strain on the patient’s family, leading in some cases to economic problems, divorce, and even suicide in the family.(33) Complications of this type are uncommon in families with an AD patient. FTD patients tend to become inattentive and careless and a danger to traffic. Changes in drinking behaviour are sometimes reported. The patient starts to drink more frequently and in larger quantities than before. The changes of behaviour, which may lead to misdiagnosis of alcohol-induced dementia, can often be controlled by a firm attitude from relatives.
Affective symptoms
The FTD patient becomes emotionally shallow and blunt, showing less concern about family and friends. The patient is described as egocentric, rigid, and lacking empathy. The early emotional changes may be difficult to differentiate from non-organic personality disorders and affective disorder. Mood changes towards euphoria, especially when associated with press of speech and overactivity, may at first be mistaken for a hypomanic or manic state. Slowly developing apathy, in combination with sparse mimical movements and verbal aspontaneity, may be misdiagnosed as depression. During the depressive reactions, which are mostly of short duration, the patient may become dysphoric, and dwell on suicidal thoughts. FTD patients are often diagnosed as depressed and treated with antidepressant medication during the early stage of the disease.(32)
Only gold members can continue reading. Log In or Register to continue