General Neuropathology of Epilepsy
Michael A. Farrell
Ingmar Blümcke
Negar Khanlou
Harry V. Vinters
Introduction
The history of the study of structural brain changes in patients with intractable epilepsy is characterized by continuing debate concerning the primacy of neuropathologic abnormalities in seizure genesis in temporal lobe epilepsy (TLE) (see Chapter 13), and intractable seizure disorders in infants and children,4,156,157 and subtle cortical cytoarchitectural alterations in primary generalized epilepsy.113,119 This chapter will focus on neocortical pathologic abnormalities and will rely for the most part on studies using human tissue. Specific structural alterations in brain tissue, which may serve as a final common pathway for the development of many diverse epilepsy-associated conditions, are discussed. Neuropathologic changes that may be attributed to epilepsy, including the neuropathologic consequences of status epilepticus (SE), are also discussed. Characteristic neuropathologic conditions that result in epilepsy syndromes (excluding developmental malformations but including epilepsy-associated neoplasms) are described.
Are There Unique Morphologic Substrates That Subserve Intrinsic Epileptogenicity in Human Neocortex?
Advances in epilepsy neuroimaging53,54,144,158 have created new challenges for the diagnostic neuropathologist, but also provide a unique opportunity to better characterize the nature of the epileptogenic lesion. A key issue in the investigation of patients with lesional epilepsy is to determine the relative contribution of the lesion and of the surrounding perilesional brain tissue to seizure genesis. Virtually all epilepsy-associated lesions may also occur in the absence of epilepsy. What, then, distinguishes the lesion that has caused epilepsy from the one that has not? Is there an underlying genetic basis for epilepsy susceptibility in such patients, and/or are there unique structural changes present in the perilesional brain tissue that provide the pathologic substrate for seizure generation? There is good evidence that some lesions are of themselves intrinsically epileptogenic; in this regard dysplastic cortical tissue is hyperexcitable.34,110 However, hyperexcitability alone is insufficient for seizure genesis. None of the epilepsy lesions has ever been shown to demonstrate spontaneous depolarization using current patch clamp techniques in an in vitro environment. Furthermore, it is clear that some lesions such as cavernous angiomas cannot be intrinsically epileptogenic, and in such cases alterations in the surrounding cortex will almost certainly provide the key to understanding seizure genesis.178
It is useful to consider a number of definitions that relate to the epileptogenic lesion and surrounding brain tissue. The lesion itself may or may not contain functionally active neural tissue. The epileptogenic zone is the area located immediately adjacent to the lesion, which is essential for seizure genesis and whose removal is generally considered to be essential for improved seizure control.17 Usually, but not always, the epileptogenic zone includes the seizure onset zone. Ideally, the functional deficit zone, which by modern neuroimaging techniques is shown to have functional abnormalities during seizure development, overlaps with the epileptogenic zone, though again this may not always be the case. Ictal symptoms result when an area of cortex is activated during epileptiform discharge and is referred to as the symptomatogenic zone. Interictal spikes utilized in the past for localization purposes reflect activity in an irritative zone, which may or may not be included within the epileptogenic zone. Finally, there is a defined area from which the seizure typically begins, referred to as the seizure onset zone. Ideally, the area in the immediate vicinity of the epileptogenic lesion would include all of the above zones, though seldom does this occur in practice. Spectacular advances in neuroimaging have led to a situation whereby the pathologist in receipt of tissue originating from epilepsy surgery can now be more certain that the resected material includes the epileptogenic zone. Additional techniques such as magnetoencephalography (MEG) have been used to map the extent of the epileptogenic zone surrounding relatively inert epilepsy-inducing lesions such as cavernomas.92,124
In attempting to determine if there is a specific pathologic change in the epileptogenic zone, immediate difficulties are encountered in terms of procuring adequate control tissue. Comparison of perilesional tissue from patients without epilepsy with the perilesional epileptogenic zone from patients with epilepsy, both having similar underlying lesions, would go a long way toward resolving the nature of any epilepsy-induced brain changes. Rarely has this been achieved. Nevertheless, new developments in molecular and pathologic techniques have provided a unique opportunity to answer questions regarding specific structural changes in the epileptogenic zone.40,41,118,121,182 Standard immunocytochemical techniques have now been supplemented with a bewildering array of sophisticated analytic techniques capable of probing the genetic and neurochemical profile of individual neurons within an epileptogenic zone. Laser capture microscopy facilitates analysis of individual neurons both in terms of neurochemical profile and surface receptor expression. Coupled with microarray studies of gene expression, it is now possible to build a very accurate genetic profile of individual neurons arising within the epileptogenic zone.27 Additionally, in vitro slice current-clamp recordings have been obtained from neurons in dysplastic brain tissue.139
Any morphologic approach to the study of the epileptogenic zone must go some way to explaining the key neurophysiologic
events in seizure genesis, namely, the recruitment and repetitive and sustained synchronous discharge of groups of neurons. Since the first Golgi studies, there has been awareness that epileptogenesis is associated with altered synaptic morphology in groups of neurons. More recently, the concept of an intrinsic deficit in neuronal function has been confirmed by the demonstration of genetically determined alterations in structural and functional properties of neuronal ion channels.64,170
events in seizure genesis, namely, the recruitment and repetitive and sustained synchronous discharge of groups of neurons. Since the first Golgi studies, there has been awareness that epileptogenesis is associated with altered synaptic morphology in groups of neurons. More recently, the concept of an intrinsic deficit in neuronal function has been confirmed by the demonstration of genetically determined alterations in structural and functional properties of neuronal ion channels.64,170
Decreased Inhibition–Increased Excitation
Morphologic studies to date have focused on the demonstration of decreased inhibition or increased excitation in the epileptogenic zone. At the very least the concept of decreased inhibition (γ-aminobutyric acid [GABA]-ergic) is simplistic and does little to elucidate the sequential change that undoubtedly take place in the epileptogenic zone as seizures become more chronic, more prolonged, and generally more severe. Attempts to determine the severity of loss of inhibitory interneurones108,120 coupled with the extent of increased excitation, resulting perhaps from axonal sprouting leading to new facilitated excitatory pathways,135 is perhaps beyond the resolution of even the sophisticated neuropathology techniques outlined above. Probing more deeply, it is difficult to see how alterations in receptor density expression on individual neurons might be used to define the origin of an epileptic focus, especially as evidence for the existence of intrinsically epileptic neurons is still not completely convincing.76,93,111,126,149 In an individual epileptogenic zone it may well be that very subtle alterations in inhibition combined with reorganization of excitatory circuits (glutamatergic) will be sufficient to result in epileptogenesis. The complexity of the decreased inhibition–increased excitation concept is illustrated by studies on chandelier cells and other cortical inhibitory interneurons in epileptogenic brain tissue, in which there is considerable variation of results within and between individual cases.6,48,49,71,88
The Neocortex in Temporal Lobe Epilepsy
Human neocortical neurons can, in response to stimulation, generate bursts of action potentials, which are caused by activation of N-methyl-D-aspartate (NMDA) receptors.13,14,65 Neurons adjacent to cavernous angiomas show spontaneous excitatory and inhibitory synaptic events and, following stimulation, demonstrate multiple action potentials.178 Spontaneous inhibitory potentials related to GABA receptor activation may also be demonstrated in human brain slices.12,15,165 Pharmacologic manipulation of receptors can lead to generation of epileptiform activity in resected neocortex, but receptor blockade or stimulation in isolation is often insufficient—the addition of an electrical stimulus being required to generate epileptiform discharges.52 Nevertheless, there is increasing evidence that synchronization, a key element in the generation of epileptiform discharges, is in part mediated by GABA receptor–mediated potentials in epileptogenic neocortex.43
Cortical Dysplasia (CD)
The spectrum of cortical dysplasia (CD)128 almost certainly extends beyond the classic description of Taylor et al., in which a combination of balloon cells and large malorientated neurons with coarse Nissl substance were the defining pathologic features.166 Detailed morphometric studies of human temporal neocortex obtained from patients undergoing resective surgery for TLE has shown a significant increase in mean neuronal somal volume in neocortical and ectopic white matter neurons.26 This subtle form of CD is not recognizable without recourse to morphometry. It is reasonable to assume that the neurophysiologic properties of classic “Taylor type” CD might also be present in subtle CD.126 For many years it has been recognized that CD tissue shows a general decrease in density of inhibitory neurons and an increase in excitatory neurons. However, detailed quantitative analysis of CD demonstrates considerable variation in the distribution and severity of GABAergic system alterations both within and between cases.3,30,64,159,167 Additionally, these studies have demonstrated considerable variation in the number of synapses per neuron both between different cases and within individual cases. The difficulty with all of these studies is a determination of the contribution of the epileptic activity itself to alterations in synaptic density, especially in terms of functional reorganization that might contribute to new synapse formation.
Intracellular recordings from the giant neurons of CD (but not from the balloon cells) show evidence of hyperexcitability when depolarized, in contrast to the balloon cells, which are essentially inert.34 Additionally, these large neurons have larger voltage-gated currents than normal-appearing neurons. Furthermore, the large neurons have increased immunoreactivity for glutamergic receptor subunits such as NMDA and also show alterations in the composition of NMDA subunits when compared with normal-appearing neurons from nonepileptic tissue.33 Intriguingly, some but not all “normal” neurons from CD tissue show similar consistent variations in NMDA subunit expression. However, in spite of all the excellent work that utilizes human epileptic tissue in vitro, it has not been possible, to date, to demonstrate intrinsic spontaneous epileptiform activity.
A Role for Glial Cells in Epileptogenesis
More recently, the contribution of the astrocytic component in any epileptogenic zone has received attention.42,68 Astrocytes are functionally active and express ion channels and neurotransmitter receptors similar to neurons. Astrocytes play a key role in modulating the neuronal extracellular environment. Long believed to be a relatively inert cell type, it is now widely recognized that astrocytes, through their ability to buffer potassium, may contribute to neuronal hyperexcitability.160 In virtually all structural lesions associated with epilepsy there are increased numbers of astrocytes either within or surrounding the lesion. Astrocytes possess voltage-activated potassium [K+] channels through an inwardly rectifying K+ channel (KIR), which have a critical role in buffering potassium levels.10,78,82,151 Fluctuations in extracellular calcium and potassium levels might well be the ionic basis for conversion of regular firing neurons into burst firing neurons capable of functioning as pacemaker neurons having the ability to recruit other neurons. There is recent evidence that genetic alterations in KIR channel subunits may contribute to the development of epilepsy susceptibility.68,163 Such alterations in potassium channel function might also be due to environmental factors. Impaired clearance of extracellular potassium could result in neuronal hyperexcitability. What is required now is a comparative study of glial cell functional properties in epileptic and nonepileptic brain tissue.
In summary, although there have been spectacular advances in neuroimaging and in the array of molecular techniques available to study the epileptogenic zone, the evidence, to date, for common underlying epilepsy-specific pathologic abnormalities that distinguish the epileptogenic zone surrounding an inert epileptic lesion from nonepileptogenic tissue surrounding a similar lesion is difficult to delineate.
Common Neuropathologic Changes That Result From Epilepsy
The effects of recurrent and prolonged seizures on the developing brain and also on the fully formed adult brain require careful consideration. Before discussing whether or not specific neuropathologic changes develop as a consequence of recurrent and prolonged seizures, it is necessary to examine whether or not there are recognizable clinical deficits that occur as a consequence of seizure activity. Intuitively, all clinicians involved in the care of patients with epilepsy suspect that frequent and/or prolonged seizures amounting to status epilepticus (SE) are harmful to the developing and mature brain. Gradual decline in school performance and behavior is frequently attributed to the effects of recurrent seizure activity in children. However, a careful examination of the literature reveals a paucity of detailed long-term prospective studies that attempt to correlate seizure frequency over many years with objective measures of cognitive function. Nevertheless, in a critical review of the studies to date (however imperfect), a statistically significant relationship was confirmed between the number of seizures or the presence of SE and decreased scores on cognitive performance.50 The effect on cognitive performance was greater for patients having uncontrolled seizures of the generalized tonic–clonic type. A similar effect was not seen with partial epilepsy. When neuroimaging findings are included, it has been shown that brain atrophy in temporal lobe epilepsy as measured by quantitative magnetic resonance imaging (MRI) is the primary mediator of the effect of epilepsy duration on neuropsychological morbidity.127
In relation to the specific issue of intellectual decline following SE in adult patients, the problem is more difficult to resolve. Significant memory impairment may occur as a result of SE, though in the majority of instances this is not permanent. Frequently SE is a reflection of underlying cerebrovascular or other serious systemic disease, the presence of which more likely determines the patient’s subsequent intellectual and cognitive performance. In a series of 1,685 patients with epilepsy who underwent comprehensive neuropsychological testing, comparison of IQs and Wechsler Adult Intelligence Scale (WAIS) scores between those who experience SE and a control group who did not, failed to demonstrate any significant intellectual decline following SE.1 Although the study may be criticized on the suitability of the WAIS-R test for measuring all cognitive functions, particularly amnesia and memory, it was not possible to demonstrate any long-lasting intellectual dysfunction.
Neuroimaging Studies
Before attempting to determine if epilepsy causes seizure-induced (recurrent unprovoked or SE) permanent neuronal injury, several confounding factors must be considered, including the neurotoxicity of antiepileptic drugs, the neuropathologic consequences of any systemic illness associated with epilepsy, any brain injury sustained during seizure episodes, and the initial precipitating event such as a prolonged febrile seizure, together with the nature of the underlying process that may have caused the epilepsy to develop in the first place. It is immediately obvious that human pathologic studies are unlikely to be able to tease out these interdependent variables. Longitudinal prospective imaging studies can readily detect alterations in the volumes of specific regions such as the hippocampus and the neocortical ribbon. Surprisingly, and although in their infancy, such studies have shown that progressive brain atrophy is not an inevitable accompaniment of epilepsy and that when volume loss does occur, it does so independently of seizure frequency and antiepileptic drug usage.101 In fact, it is felt that any developing atrophy is more likely to be due to a combination effect of normal aging and the underlying disease process that caused the epilepsy to occur in the first place. It must be stressed that such studies are difficult to execute and will require large numbers and several years of follow-up.
Patients with temporal lobe epilepsy demonstrate mean reductions of from 4% to 6.6% in cerebellar volume when compared with nonepileptic control patients; in general, a longer duration of TLE with an increased lifetime number of generalized tonic–clonic seizures were present in those with cerebellar atrophy. Furthermore, the cerebellar atrophy appeared to occur independently of cerebral cortical atrophy.75
Recent neuroimaging studies, especially those which measure water diffusion through the brain, have demonstrated alterations in diffusivity after both prolonged seizure activity and single seizures. Single seizures may be followed by focal changes in diffusion-weighted imaging (DWI), which invariably return to interictal levels within a few hours.147 The pathologic correlate of the altered DWI is as yet unknown, though it is likely that transient edema may occur. Alterations in DWI that have developed following prolonged seizure or SE79 have gradually returned to interictal levels, though in a few instances the changes in DWI have been followed by brain atrophy.96 SE with minor or no motor disturbance (i.e., nonconvulsive SE), either generalized or focal, may be associated with MRI changes.37 Thus, although the neuroimaging evidence for seizure-induced neuronal injury is beginning to accumulate, currently it is difficult to be certain of the long-term significance of these changes in terms of clinically apparent brain injury.
Human Pathologic Studies
In a few well-documented human pathologic studies, it has been possible to demonstrate neuronal injury that has clearly occurred as a direct result of seizure activity. In a detailed imaging and autopsy study, a 28-year-old man was shown—during 5 months of idiopathic convulsive SE—to develop evolution of generalized cerebral cortical atrophy with severe neuronal loss and gliosis. The patient had not become hypoxic or hypotensive in the course of the illness.132 In another report, a 9-year-old girl with refractory focal SE developed progressive generalized brain atrophy over a 3-year period during which the MRI showed progression from normal through increased focal T2 signal to generalized atrophy. Brain biopsy demonstrated neuronal loss and gliosis.51 Again, there was no evidence of any confounding variable such as hypotension or hypoxia. Crossed cerebellar atrophy has been demonstrated by MRI,94,116 positron emission tomography (PET),85 and pathologically97,164 in the cerebellar hemisphere contralateral to the cerebral hemisphere from which unilateral persistent seizure activity arose, thereby providing good evidence for enhanced neuronal activity, transmitted via corticocerebellar pathways, as the likely explanation for neuronal injury.
Although the neuropathologic consequences of SE in humans have not been extensively studied in recent years,154,175 there is good evidence from autopsy examinations that widespread neuronal necrosis with varying degrees of gliomes-odermal proliferation may occur,103 though not invariably so, after SE.38 There is general agreement that the severity of such neuronal injury is less in children than in adults.70 The hippocampus shows the most striking changes, with widespread neuronal necrosis. Similar changes may be present in the thal-amus and in a patchy distribution throughout the neocortex. The extent of any associated astrogliosis or microglial cell proliferation depends, to a large extent, on the duration of a given patient’s survival following SE. Repeated episodes of SE could result in sufficiently severe degrees of neuronal loss to eventually lead to cortical atrophy.
Controversy has arisen in relation to watershed/borderzone ischemic lesions occurring in SE. In a review of the neuropathologic consequences of SE,28 which cited the classic work of
Corsellis and Bruton, it was suggested that watershed distribution necrosis is feature of SE. In subsequent correspondence, Bruton29 explicitly stated that such lesions were not described in his original publication. In reply, the editors cited incorrect referencing, declaring that the description of watershed lesions in SE should have been credited to the classic work of Norman,123 originally published in 1966 and reprinted in 1987. Unfortunately, however, a very careful scrutiny of the original 1966 work and of the subsequent 1987 reprint has failed to locate any reference whatsoever to watershed lesions in the brains of patients dying of SE. It is our belief that neuronal necrosis having a watershed distribution cannot be attributed to SE alone and that some other contributory factor, such as profound hypotension, should be sought, either as the cause of or a complication of SE.
Corsellis and Bruton, it was suggested that watershed distribution necrosis is feature of SE. In subsequent correspondence, Bruton29 explicitly stated that such lesions were not described in his original publication. In reply, the editors cited incorrect referencing, declaring that the description of watershed lesions in SE should have been credited to the classic work of Norman,123 originally published in 1966 and reprinted in 1987. Unfortunately, however, a very careful scrutiny of the original 1966 work and of the subsequent 1987 reprint has failed to locate any reference whatsoever to watershed lesions in the brains of patients dying of SE. It is our belief that neuronal necrosis having a watershed distribution cannot be attributed to SE alone and that some other contributory factor, such as profound hypotension, should be sought, either as the cause of or a complication of SE.
Common Brain Lesions Associated with Epilepsy
In this section, selected features of several characteristic neuropathologic entities associated with seizure disorders are described. Structural features of these lesions (and their relationship to epilepsy) will be emphasized and selectively illustrated. Especially in patients with temporal lobe epilepsy, “dual pathology” (e.g., mesial temporal/hippocampal sclerosis together with cortical dysplasia or a cortical neoplasm) is not infrequently encountered.146
Neoplasms
Both primary and secondary (metastatic) neoplasms of the central nervous system (CNS) are characterized by their distinctive histologic appearance, location(s), and immunohistochemical phenotype. Preoperative neuroimaging studies, including magnetic resonance spectroscopy (to look for levels of N-acetylaspartate, choline, and creatine, as well as choline:creatine ratios) may be helpful in predicting how aggressive a neoplasm is likely to be.57 The predicted biologic behavior of primary brain tumors is reflected in how they are categorized according to the World Health Organization (WHO) classification system.90,91 When astrocytic tumors are considered as a major subset of all infiltrating glial neoplasms (gliomas), their degree of malignancy is readily ascertained by examining distinctive histologic features present in the tumor—characteristics that can quite accurately be evaluated in a simple hematoxylin-eosin–stained tissue section.46 Unfortunately, these grading parameters appear to be less useful in evaluating CNS tumors in the pediatric age group.66 However, the neuropathologic diagnosis and study of brain neoplasms is now being rapidly refined using tools of immunohistochemistry and molecular genetics.112 These methodologies will almost certainly supersede evaluation of the subjective or semi-objective histologic criteria upon which tumor diagnosis is now based, in terms of their predictive value. Immunohistochemical techniques can detect evidence of tumor differentiation along astrocytic and/or neuronal lines, or (in the case of a metastasis) suggest a site of origin of the neoplasm, even when this is not apparent on initial workup of a given patient. Assessment of the “proliferative potential” of tumor cells can now be undertaken by using one or more several straightforward immunohistochemical markers (e.g., anti-Ki-67), many of them effectively used in paraffin sections,133,134 thereby allowing for meaningful studies on archival material. Without question, modifications of the histopathologic approach to classify CNS neoplasms have also resulted from recent molecular-genetic studies. Further advances in the “molecular pathology” of CNS neoplasia are likely to revolutionize how we categorize and treat gliomas, which remain highly refractory to even the “best” medical therapy. Association between allelic losses and sensitivity to polychemotherapy in oligodendrogliomas58,63,81,171 or epigenetic regulation of D-6-methylguanine-DNA methyltransferase (MGMT) with better responsiveness to temozolomide are hallmarks of this progress.74 Even tumors that we have in the past “lumped together” (e.g., using the term glioblastoma) are now known to have distinctive molecular signatures (defined by patterns of expression of epidermal growth factor receptor [EGFR] components and PTEN) that predict how these neoplasms are likely to respond to EGFR kinase inhibitors.114 Highly individualized therapy for primary brain tumors is likely to result from such advances. Further analysis of the complex molecular genetic interface between familial brain tumor syndromes (e.g., neurofibromatosis) and neurocutaneous disorders/phakomatoses is rapidly evolving.95,102,103,112
The study of neoplasms associated with epilepsy, which in general tend to behave in an anomalous (and usually fairly benign) fashion, by means of these new cellular and molecular tools will be of particular interest. However, it is important to remember that any intra-axial neoplasm, in particular one with a cortical (especially hippocampal) component (Fig. 1), can cause seizures, as can any extra-axial tumor that significantly compresses, displaces, or invades the cerebral hemispheres. In one large series of almost 500 patients with brain metastases, seizures were noted at the time of presentation or at some point during the course of the illness in over 25% of individuals. Seizure likelihood was highest with melanoma (occurring in two thirds of affected individuals), while tumors that had spread to the CNS from lung, breast, or an unknown primary were also a common cause of seizures.55 Seizures have been estimated to occur in 80% of low-grade (2 out of 4) glioma patients, in almost one third of those with high-grade glioma, 20% of those who harbor a meningioma, and one tenth of patients with primary CNS lymphoma.77 The pathophysiology of tumor-related seizures is multifactorial and extremely complex (for an excellent topical review, see reference 148). Important factors that contribute to tumor-related seizures may include a relative loss of inhibitory synapses or inhibitory interneurons in tumor-adjacent tissue, subtle pH alterations (peritumoral pH is slightly alkaline), ion level changes (e.g., decreased extracellular Mg2+), amino acid fluctuations (e.g., affecting alanine, phosphoethanolamine, and glutamine), enzymatic derangements and increased excitatory activity in peritumoral tissue secondary to abnormal glutamatergic transmission associated with altered receptor subunit expression, and abnormal intercellular communication resulting from derangements in gap junction proteins (e.g., connexins CX43 on astrocytes, CX32 on oligodendroglia, and CX26 and CX32 on neurons).
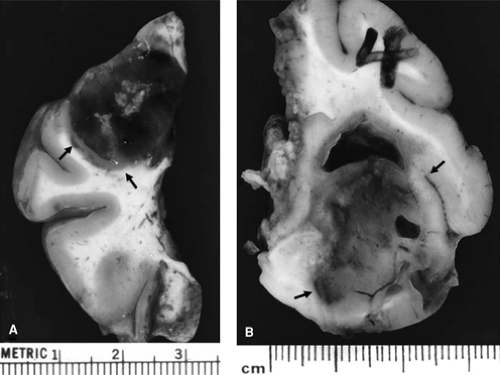 FIGURE 1. Tumors associated with intractable epilepsy, resected from two different patients. Note clear demarcation of the tumors from surrounding brain (indicated by arrows). Tumor shown in panel A is significantly hemorrhagic, whereas neoplasm in panel B is relatively homogeneous with focal cavitation. Both tumors clearly involve the cortex. (See the color insert.) |
Table 1 Brain Tumors Associated with Epilepsy | |||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||
The proportion of tumors seen among resected specimens from epileptic patients varies tremendously among epilepsy surgery centers, depending obviously on the interests and expertise of local clinicians as well as referral patterns to a given institution. In large series describing the neuropathologic features of temporal lobe resections carried out for pharmacoresistent seizures, neoplasms account for 25% to more than 50% of the resultant specimens.104,105,131,179,180,181 Some tumors (see below) may coexist with foci of cortical disorganization or dysplasia. In patients with pharmacoresistent TLE, dual and even multiple lesions may be encountered (e.g., hippocampal sclerosis together with malformations of cortical development and/or low-grade neoplasms).56 The range of tumors encountered in patients with TLE is variable and includes the full spectrum of primary brain tumors, although gangliogliomas, pilocytic astrocytomas, and dysembryoplastic neuroepithelial tumors (WHO grade I tumors) are unusually common.179,180,181 Extratemporal resections for intractable epilepsy also often yield neoplasms, usually of low histologic grade and having histopathologic features comparable to those encountered in
the temporal lobes. All studies—in both adults and children—emphasize the remarkably high likelihood of patients being seizure free (i.e., an Engel class I outcome) after tumor resection, even when only a “lesionectomy” is performed.31,67 The Bonn group, with one of the world’s largest experiences of epilepsy-associated neoplasia, has emphasized that biologically aggressive infiltrating glial tumors or tumors with a ganglion cell component (astrocytoma or ganglioglioma WHO grade III) are distinctly unusual among tumor resections performed for intractable seizure disorders.104 The biologic behavior and appearance of some of the specific tumor types commonly encountered are worth reviewing in greater detail (see Table 1).
the temporal lobes. All studies—in both adults and children—emphasize the remarkably high likelihood of patients being seizure free (i.e., an Engel class I outcome) after tumor resection, even when only a “lesionectomy” is performed.31,67 The Bonn group, with one of the world’s largest experiences of epilepsy-associated neoplasia, has emphasized that biologically aggressive infiltrating glial tumors or tumors with a ganglion cell component (astrocytoma or ganglioglioma WHO grade III) are distinctly unusual among tumor resections performed for intractable seizure disorders.104 The biologic behavior and appearance of some of the specific tumor types commonly encountered are worth reviewing in greater detail (see Table 1).
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


