The successful use of psychotropic drugs demands an understanding of their pharmaceutical, pharmacokinetic, and pharmacodynamic properties.
♦ Pharmaceutical properties: Pharmaceutical formulations can be manipulated to produce different durations of action, for example the use of oily emulsions of antipsychotic drugs in depot formulations.
♦ Pharmacokinetic properties: Pharmacokinetics is the mathematical description of the disposition of drugs in the body by absorption, distribution (to plasma proteins and tissues), and elimination (usually by hepatic metabolism and renal excretion). Differences in drug disposition determine differences in dosage regimens and are important for drug interactions.
♦ Pharmacodynamic properties: Pharmacodynamics is the study of the pharmacological actions of drugs and how actions at the molecular level are translated, via actions at cellular, tissue, and organ levels, into therapeutic or adverse effects. The known pharmacological actions of psychotropic drugs are not necessarily the actions that produce their therapeutic or adverse effects.
Dosage regimens
A drug dosage regimen is a recipe for drug administration, intended to produce the desired therapeutic effect with a minimum of unwanted effects. It is described in terms of the pharmaceutical formulation, the dose, and the frequency and route of administration used. The duration of administration is also important.
In treating any condition, it is best to learn initially to use a few drugs, preferably well-established ones, and to expand one’s repertoire with increasing experience.
The choice of drug depends firstly on the indication—obviously an antidepressant will be the drug of choice for a patient with depression, if drug therapy is thought to be required. The choice of antidepressant will depend on features of the disease and other factors. For example, some antidepressants are more sedative and anxiolytic than others, and can be helpful in patients who are agitated. The avoidance of adverse effects or interactions can also dictate the choice; for example, tricyclic antidepressants should be avoided in men with prostatic hyperplasia and selective serotonin reuptake inhibitors (SSRIs) should not be used in children, because of the increased risk of suicidal ideation.
It is usual to start therapy with published dosage recommendations, generally beginning at the lower end of the recommended dosage range and monitoring for a therapeutic effect. A common error is to give a starting dose of a drug and then to add or substitute another drug if the first does not work. This is usually bad practice. If the desired effect does not occur with the initial dosage, increase the dose gradually until the effect occurs or the upper limit of the recommended range is reached (although adverse effects may limit this process). Only then should another drug be tried. Sometimes a poor response is due to poor adherence to therapy; careful explanation of the condition and the need for therapy helps.
Psychotropic drugs can be given orally or parenterally, and as immediate-release or modified-release formulations. Most drug administration is oral, but parenteral therapy can be useful to guarantee administration (e.g. depot formulations in schizophrenia) and for a quicker onset of action (e.g. in the treatment of acute mania). Modified-release products are used for long-term therapy. They can be given less often and produce a smoother profile of blood concentrations (Fig. 6.2.1.1).(1) The advantage of intramuscular modified-release (depot) formulations of antipsychotic drugs is that drug delivery can be ensured by supervised infrequent administration (say every 2 weeks). Different modified-release formulations of the same compounds have different release characteristics and are not interchangeable; for example, when prescribing a modifiedrelease oral formulation of lithium always give the patient the same formulation and specify the brand name on the prescription.
Combination formulations (e.g. a phenothiazine plus a tricyclic antidepressant in a single tablet) do not allow flexibility of prescribing and should generally be avoided. Important exceptions include combination analgesic formulations (e.g. co-codamol, which contains paracetamol plus codeine) and combinations of levodopa with a dopa decarboxylase inhibitor (benserazide or carbidopa).
Treatment of children
There are no uniform rules for determining dosage regimens in children. Pharmacokinetics and pharmacodynamics are different for some drugs but not others.
♦ Absorption is not greatly different from absorption in adults.
♦ The distribution of water-soluble drugs is different, but psychotropic drugs are lipid-soluble.
♦ Protein binding is reduced in neonates; phenytoin is affected.
♦ Hepatic oxidative metabolism and glucuronide conjugation are deficient in neonates, and mature at variable rates; this is important for psychotropic drugs.
♦ Glomerular and renal tubular functions are immature in neonates and take about 6 months to reach adult values.
If a child needs a psychotropic drug, consult the manufacturer’s literature and always start with a low dosage.
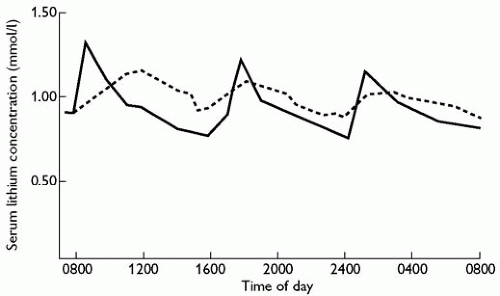
Fig. 6.2.1.1 Administration of lithium in immediate-release and modified-release oral formulations. The immediate-release formulation (solid line) produces rapid peaks of serum concentration and large fluctuations during a dosage interval. In contrast, the modified-release formulation (dotted line) is more slowly absorbed but produces much less fluctuation in serum concentrations. Note also that the apparent half-life of lithium is longer after administration of the modified-release formulation; this is not the true half-life of lithium, but the half-life of its release from the modified-release formulation. (Adapted from A. Amdisen, Variation of serum lithium concentration during the day in relation to treatment control, absorptive side effects and the use of slow-release tablets, Acta Psychiatrica Scandinavica, 207, 55-7, copyright 1969, John Wiley & Sons, Inc.)
Treatment of elderly people
Pharmacokinetic differences in old age are more predictable than in children, but pharmacodynamic changes are variable.
♦ Absorption is not greatly affected.
♦ Elderly people have less body fat, and so lipid-soluble drugs may be more highly concentrated in the brain; however, this effect varies unpredictably from drug to drug (e.g. the apparent volume of distribution of diazepam is increased while that of nitrazepam is not).
♦ Protein binding is reduced in elderly people; phenytoin is affected.
♦ Hepatic metabolism is reduced in frail but not in fit old people; this effect is proportional to liver size.
♦ Renal function is impaired with age; use creatinine clearance, measured or estimated (not eGFR), as a guide.
♦ Inappropriate polypharmacy is common in old people, increasing the risk of drug interactions.
When treating an elderly person with a psychotropic drug always start with a low dosage and increase dosages more slowly.
Pregnancy and breast feeding
Anticonvulsants are teratogenic.(2) For example, sodium valproate has been associated with spina bifida, cardiac malformations, hypospadias, anomalies of the brain and face, coarctation of the aorta, and limb reduction defects.(3) Few other psychoactive drugs are teratogenic. However, most of them cross the placenta and some can cause withdrawal symptoms in the neonate. The teratogenicity of lithium has been overstated in the past; the main risk is cardiovascular teratogenicity, but although the risk of Ebstein’s anomaly is increased, the absolute risk (0.05-0.1 per cent) is still very small;(4) nevertheless, some advise that it should be avoided or used with caution in the first trimester of pregnancy,(5) and fetal sonography is recommended at 18-20 weeks after first-trimester exposure.(3)
Although most psychoactive drugs are lipid-soluble and therefore enter the breast milk, few do so in high enough amounts to trouble the neonate; if a neonate becomes drowsy while breast feeding, reduce the mother’s dosage or stop breast feeding. Lithium appears in the breast milk and can be found in the serum of breast-fed babies in variable concentrations, up to half of those in the mother. Because neonates have immature renal function, some recommend avoiding breast feeding.(6) However, others consider that the benefits of breast feeding to mother and child outweigh the small risk of lithium toxicity.(4) The following advice has been given:(3)
♦ Educate the mother about the manifestations of toxicity.
♦ Explain the risks of dehydration.
♦ Consider partial or total formula supplements during episodes of illness or dehydration.
♦ Suspend breast feeding if toxicity is suspected.
♦ Check infant and maternal serum concentrations.
Pharmacokinetics—drug disposition
Most psychotropic drugs are rapidly and well absorbed after oral administration. However, drugs can be removed by various processes before they reach the systemic circulation. The fraction of drug that reaches the systemic circulation is called its systemic availability (or, more commonly, bioavailability).
After oral administration a formulation will generally disintegrate in the stomach and the drug it contains will dissolve in gastric contents. However, drugs are not generally absorbed in the stomach. After gastric emptying they are for the most part absorbed in the jejunum and ileum, and some are absorbed from the colon as well. During transit across the gut wall they may be metabolized by an oxidative isozyme of cytochrome P450, CYP3A4, and can be secreted back into the gut lumen by P glycoprotein. When they enter the portal circulation they may be eliminated by the liver. If hepatic metabolism is extensive, a large amount of drug will be removed during this first passage through the liver. For example, clomethiazole has extensive first-pass metabolism in the liver and its systemic availability is low (about 40 per cent); thus, intravenous doses are considerably lower than oral doses. In severe liver disease, such as cirrhosis, or when there is arteriovenous shunting, this presystemic metabolism is reduced and the systemic availability increases up to 90 per cent; oral doses of clomethiazole should be reduced in liver disease.(7)
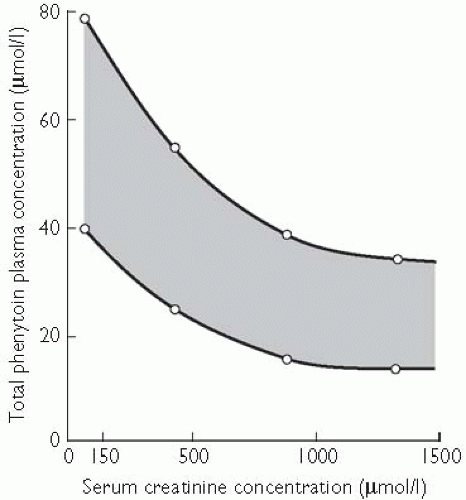
In the systemic circulation drugs are bound to plasma proteins and distributed to the tissues. Protein binding is important for drugs that are highly bound (over 90 per cent) and not widely distributed to the body tissues; in those cases protein-binding displacement can result in a large rise in the amount of unbound drug available to the target tissue. This is important for phenytoin, which is 90 per cent bound to plasma albumin and has a low volume of distribution. The binding of phenytoin is reduced when the serum albumin concentration falls (in chronic liver disease, the nephrotic syndrome, protein malnutrition, or the third trimester of pregnancy), when binding to the protein is abnormal (in chronic renal insufficiency), or when another drug (e.g. sodium valproate) causes displacement. Acute displacement causes phenytoin toxicity, but only transiently, because in the case of phenytoin an increase in unbound concentration causes it to be more rapidly eliminated. When measuring plasma phenytoin concentrations in patients in whom protein binding is reduced, the target concentration (and the laboratory will measure total drug, i.e. bound plus unbound) is reduced (see Fig. 6.2.1.2).
In chronic renal insufficiency the protein binding of phenytoin is reduced. This leads to an increase in the unbound plasma (or serum) concentration relative to the total concentration; the target total concentration therefore falls. The shaded area shows the range of plasma phenytoin concentrations that one would generally aim to achieve (the target concentration range) in treating a patient with epilepsy. As renal function deteriorates (indicated here by an increase in serum creatinine concentration), the target range for plasma phenytoin concentration falls from 40-80 µmol/l when renal function is normal to 10-30 µmol/l in severe renal insufficiency.
After absorption and distribution most psychoactive drugs are cleared from the body by hepatic metabolism;(8) impaired liver function, if severe (for the liver has a large capacity), reduces their elimination, and dosages should be reduced.
Lithium is cleared solely by renal elimination and therefore the dosage should be reduced in proportion to the creatinine clearance. Since renal function falls with age, lithium dosages should be lower in older people.(9)
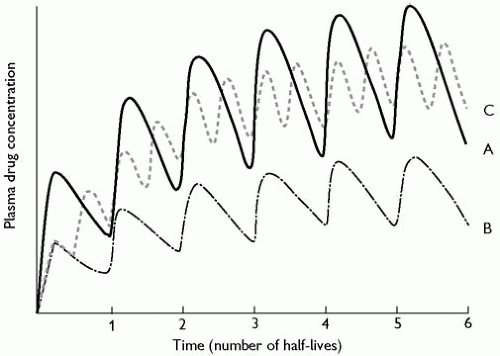
The half-life of a drug is a function of its clearance and its distribution volume: the slower the rate of clearance or the more extensive the distribution the longer the half-life. If a drug is given in a regular maintenance dose, the amount of drug in the body will gradually accumulate; however, as the amount in the body increases, the rate at which it is eliminated also rises, and eventually a plateau (or steady state) is reached when the amount eliminated during a dosage interval equals the dose (the maintenance dose). The time it takes to reach this steady state depends on the half-life of the drug; about 94 per cent of the steady-state value will be reached after four half-lives (Fig. 6.2.1.3, curve A). For example, lithium has a half-life of about 24 h; after 4 days of maintenance therapy with the same regular dose a steady state will be reached; this does not depend on the dose or frequency of administration (Fig. 6.2.1.3, curves B and C). If a modified-release formulation is used and the half-life of absorption of the drug from the formulation is longer than the drug’s own half-life, the longer (apparent) half-life will determine the time to steady state; for example, the apparent half-life of flupentixol after the administration of flupentixol decanoate is 17 days, compared with 36 h for flupentixol after oral administration. When using depot antipsychotic drugs, which have long half-lives of absorption, steady-state therapy should first be established with an ordinary formulation.
Fig. 6.2.1.2 In chronic renal insufficiency the protein binding of phenytoin is reduced. This leads to an increase in the unbound plasma (or serum) concentration relative to the total concentration; the target total concentration therefore falls. The shaded area shows the range of plasma phenytoin concentrations that one would generally aim to achieve (the target concentration range) in treating a patient with epilepsy. As renal function deteriorates (indicated here by an increase in serum creatinine concentration) the target range for plasma phenytoin concentration falls, from 40-80 µmol/l when renal function is normal to 10-30 µmol/l in severe renal insufficiency (I. Odar-Cederlöf, unpublished data.)
Fig. 6.2.1.3 Curve A—during the regular administration of a maintenance dose of a drug the amount of drug in the body rises after a dose, reaches a peak, and then falls as the drug is distributed to the tissues and eliminated. If another dose is given soon after the first, the plasma concentration will rise by the same amount as before but will fall faster after peaking, since most drugs obey first-order kinetics and the plasma concentration falls exponentially. Thus, when a drug is given repeatedly the mean plasma concentration rises more slowly with each successive dose, until eventually a steady state is reached, when the amount eliminated in a dosage interval is equal to the dose itself. This takes about four half-lives of the drug. Curve B represents the concentrations during administration of half the dose given at the same frequency. The time taken to reach steady state is the same in both cases, but the eventual steady-state concentration in case B is half that in case A, being proportional to the dose. Curve C represents the concentrations during administration of half the dose given twice as often (i.e. the total dose is unchanged). Neither the time taken to reach steady state nor the eventual mean steady-state concentration is affected. However, the fluctuations in plasma concentration during a dosage interval are reduced (cf. Fig. 6.2.1.1). (Adapted from Amdisen, A. Variation of serum lithium concentration during the day in relation to treatment control, absorptive side effects and the use of slow-release tablets, Acta Psychiatrica Scandinavica, 207, 55-57. Copyright 1969, John Wiley & Sons, Inc.)
Curve A shows that during the regular administration of a maintenance dose of a drug the amount of drug in the body rises after a dose, reaches a peak, and then falls as the drug is distributed to the tissues and eliminated. If another dose is given soon after the first, the plasma concentration will rise by the same amount as before but will fall faster after peaking, since most drugs obey first-order kinetics and the plasma concentration falls exponentially. Thus, when a drug is given repeatedly the mean plasma concentration rises more slowly with each successive dose, until eventually a steady state is reached, when the amount eliminated in a dosage interval is equal to the dose itself. This takes about four half-lives of the drug. Curve B represents the concentrations during administration of half the dose given at the same frequency. The time taken to reach steady state is the same in both cases, but the eventual steady-state concentration in case B is half that in case A, being proportional to the dose. Curve C represents the concentrations during administration of half the dose given twice as often (i.e. the total dose is unchanged). Neither the time taken to reach steady state nor the eventual mean steady-state concentration is affected. However, the fluctuations in plasma concentration during a dosage interval are reduced (cf. Fig. 6.2.1.1). Kinetic characteristics of some psychotropic drugs are shown in Table 6.2.1.1.
Pharmacological actions of drugs
Psychotropic drugs interfere with neurotransmitter functions in several ways—via actions on neurotransmitter receptors, storage, release, reuptake, and metabolism. Transmembrane neurotransmitter receptors are broadly speaking of two types—ionotropic and metabotropic receptors. Ionotropic receptors (e.g. nicotinic acetylcholine, glycine, GABA, and NMDA, AMPA, and kainate receptors) incorporate ion channels in their structures and mediate rapid responses. Metabotropic receptors (e.g. G protein-coupled receptors such as adrenaline, noradrenaline, cannabinoid, dopamine, opioid, and serotonin receptors other than 5HT3 receptors) produce their effects via signal transduction systems, which activate second messengers or ion channels, and produce longer lasting responses.
Agonist action at a receptor
Agonists are substances that act by stimulating the action of a receptor.
Benzodiazepines bind to benzodiazepine receptors in the spinal cord, brainstem, cerebellum, limbic system, and cerebral cortex. These receptors are associated with receptors for the inhibitory neurotransmitter γa-minobutyric acid (GABA), linked to a chloride channel.(10) The benzodiazepines enhance the action of GABA through its chloride channel, the presumed mechanism whereby they are anxiolytic and hypnotic. Some other hypnotics that have non-benzodiazepine structures also act via benzodiazepine receptors: zopiclone binds to the GABA-benzodiazepine receptor complex, but at a site different from that of benzodiazepines;(11) clomethiazole binds to a binding site distinct from those of benzodiazepines and barbiturates;(12) zolpidem binds to a subtype of binding site called BZ1, found on GABA neurones in the sensorimotor cortex and extrapyramidal tracts.(10)
The triptans (such as sumatriptan, naratriptan, zolmitriptan), which are used to treat migraine, are agonists at 5-hydroxytryptamine (5-HT1B/D) receptors, causing vasoconstriction. They are therefore contraindicated in patients with cardiovascular disease and in those with hemiplegic or basilar migraine because of the fear of stroke.(13)
Antagonist action at a receptor
Antagonists are substances that have no actions of their own at receptors and act by preventing the action of an agonist, usually an endogenous one.
The antipsychotic (neuroleptic) drugs are all antagonists at receptors for the endogenous neurotransmitter dopamine; this is thought to be the basis of their antipsychotic actions in the mesolimbic system (via Dl and D4 receptors) and undoubtedly produces their adverse effects in the extrapyramidal tracts (via D2 receptors). The so-called atypical antipsychotic drugs (including clozapine and risperidone) have little effect on D2 receptors and less commonly cause extrapyramidal adverse effects.(14)
Table 6.2.1.1 Pharmacokinetic information about some psychotropic drugs
a CYP refers to isozymes of the cytochrome P-450 family of enzymes.
b All these drugs are partly metabolized to active metabolites, Some of the metabolites have long half-lives (e.g. diazepam is metabolized to desmethyldiazepam). Some benzodiazepines (e.g. clorazepate and prazepam) are completely metabolized to active metabolites with long half-lives.