General Principles of Psychopharmacology
For a more detailed discussion of this topic, see General Principles of Psychopharmacology, Sec 31.1, p. 2965, and Pharmacokinetics and Drug interactions, Sec 31.2, p. 2988, in Comprehensive Textbook of Psychiatry, 9th Edition.
Introduction
Since their emergence in the mid-1950s, drugs used to treat psychiatric disorders have become among the most prescribed classes of pharmaceutical agents. In addition to agents with Food and Drug Administration (FDA) approval for use as primary interventions for mental disorders, many other drugs are used “off label.” In fact, many of the chapters that follow discuss compounds that are intended to treat general medical or neurologic conditions.
There are three general terms, used interchangeably, that describe drugs that treat psychiatric disorders. These are psychotropic drugs, psychoactive drugs, and psychotherapeutic drugs. Traditionally, these agents have been divided into four categories: (1) antipsychotic drugs or neuroleptics used to treat psychosis, (2) antidepressant drugs used to treat depression, (3) antimanic drugs or mood stabilizers used to treat bipolar disorder, and (4) antianxiety drugs or anxiolytics used to treat anxious states (which are also effective as hypnotics in high dosages). Such categoric distinctions, however, have become less valid for the following reasons:
Many drugs of one class are used to treat disorders previously assigned to another class. For example, most antidepressant drugs are now also used to treat a broad range of anxiety disorders.
Drugs introduced as treatments for schizophrenia, agents such as the serotonin–dopamine antagonists (SDAs), are also indicated for the management of bipolar disorder and appear to have some antidepressant activity.
Drugs from all four categories are used to treat symptoms and disorders such as insomnia, eating disorders, behavioral disturbances associated with dementia, and impulse-control disorders.
Drugs such as clonidine (Catapres), propranolol (Inderal), verapamil (Isoptin), modafinil (Provigil), and gabapentin (Neurontin) can effectively treat a variety of psychiatric disorders and do not fit easily into the traditional classification of drugs.
Some descriptive psychopharmacologic terms are arbitrary and overlap in meaning. For example, anxiolytics decrease anxiety, sedatives produce a calming or relaxing effect, and hypnotics produce sleep. However, most anxiolytics function as sedatives and at high doses can be used as hypnotics, and all hypnotics at low doses can be used for daytime sedation.
Classification
This book uses a classification in which each drug is discussed according to its pharmacologic category. Each drug is described in terms of its pharmacologic actions, including pharmacodynamics and pharmacokinetics. Indications, contraindications, drug–drug interactions, and adverse side effects are also discussed.
Table A (see p. xiii) lists each psychotherapeutic drug according to its generic name, trade name, and chapter title and number in which it is discussed.
Pharmacologic Actions
The main determinants of the clinical effects of a drug on an individual are determined by its pharmacokinetic and pharmacodynamic properties. In simple terms, pharmacokinetics describes what the body does to the drug, and pharmacodynamics describes what the drug does to the body. Pharmacokinetic data trace the absorption, distribution, metabolism, and excretion of the drug in the body. Pharmacodynamic data measure the effects of the drug on cells in the brain and other tissues of the body.
Pharmacokinetics
Absorption
Drugs reach the brain through the bloodstream. Orally administered drugs dissolve in the fluid of the gastrointestinal (GI) tract—depending on their lipid solubility and the GI tract’s local pH, motility, and surface area—and are then absorbed into the blood.
Stomach acidity may be reduced by proton pump inhibitors, such as omeprazole (Prilosec), esomeprazole (Nexium), and lansoprazole (Prevacid); by histamine H2 receptor blockers, such as cimetidine (Tagamet), famotidine (Pepcid), nizatidine (Axid), and ranitidine (Zantac); or by antacids. Gastric and intestinal motility may be either slowed by anticholinergic drugs or increased by dopamine receptor antagonists (DRAs), such as metoclopramide (Reglan). Food can also increase or decrease the rate and degree of drug absorption.
As a rule, parenteral administration can achieve therapeutic plasma concentrations more rapidly than can oral administration. However, some drugs are deliberately emulsified in an insoluble carrier matrix for intramuscular (IM) administration, which results in the drug’s gradual release over several weeks. These formulations are called depot preparations. Intravenous (IV) administration is the quickest route for achieving therapeutic blood concentrations, but it also carries the highest risk of sudden and life-threatening adverse effects.
Distribution and Bioavailability
Drugs that circulate bound to plasma proteins are called protein bound, and those that circulate unbound are called free. Only the free fraction can pass through the blood–brain barrier.
The distribution of a drug to the brain is governed by the brain’s regional blood flow, the blood–brain barrier, and the drug’s affinity with its receptors in the brain.
High cerebral blood flow, high lipid solubility, and high receptor affinity promote the therapeutic actions of the drug.
High cerebral blood flow, high lipid solubility, and high receptor affinity promote the therapeutic actions of the drug.
A drug’s volume of distribution is a measure of the apparent space in the body available to contain the drug, which can vary with age, sex, adipose tissue content, and disease state. A drug that is very lipid soluble, such as diazepam (Valium), and thus is extensively distributed in adipose tissue may have a short duration of clinical activity despite a very long elimination half-life.
Bioavailability refers to the fraction of the total amount of administered drug that can subsequently be recovered from the bloodstream. Bioavailability is an important variable because FDA regulations specify that the bioavailability of a generic formulation can differ from that of the brand-name formulation by no more than 30%.
Metabolism and Excretion
Metabolic Routes
The four major metabolic routes for drugs are oxidation, reduction, hydrolysis, and conjugation. Metabolism usually yields inactive metabolites that are readily excreted. However, metabolism also transforms many inactive prodrugs into therapeutically active metabolites.
The liver is the principal site of metabolism, and bile, feces, and urine are the major routes of excretion. Psychotherapeutic drugs are also excreted in sweat, saliva, tears, and breast milk.
Quantification of Metabolism and Excretion
Four important parameters regarding metabolism and excretion are time of peak plasma concentration, half-life, first-pass effect, and clearance.
The time between the administration of a drug and the appearance of peak plasma concentrations varies according to the route of administration and rate of absorption.
A drug’s half-life is the amount of time it takes for metabolism and excretion to reduce a particular plasma concentration by half. A drug administered steadily at time intervals shorter than its half-life will reach 97% of its steady-state plasma concentration after five half-lives.
The first-pass effect refers to the initial metabolism of orally administered drugs within the portal circulation of the liver and is described as the fraction of absorbed drug reaching the systemic circulation unmetabolized.
Clearance is a measure of the amount of the drug excreted from the body in a specific period of time.
Cytochrome P450 Enzymes
The Cytochrome P450 (CYP) enzyme system is responsible for the inactivation of most psychotherapeutic drugs. It is so named because the heme-containing enzymes strongly absorb light at a wavelength of 450 nm. Although present throughout the body, these enzymes act primarily in the endoplasmic reticulum of the hepatocytes and the cells of the intestine. Thus, cellular pathophysiology, such as that caused by viral hepatitis or cirrhosis, may affect the efficiency of drug metabolism by the CYP enzymes.
The human CYP enzymes comprise several distinct families and subfamilies. In the CYP nomenclature, the family is denoted by a numeral, the subfamily by a capital letter, and the individual member of the subfamily by a second numeral (e.g.,
2D6). Persons with genetic polymorphisms in the CYP genes that encode inefficient versions of CYP enzymes are considered poor metabolizers.
2D6). Persons with genetic polymorphisms in the CYP genes that encode inefficient versions of CYP enzymes are considered poor metabolizers.
Table 1-1 Comparison of Metabolic Inhibition and Metabolic Induction | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
There are two mechanistic processes involving the CYP system: inhibition and induction (Table 1-1):
Induction. Expression of the CYP genes may be induced by alcohol, certain drugs (barbiturates, anticonvulsants), or smoking. For example, an inducer of CYP 3A4, such as cimetidine, may increase the metabolism and decrease the plasma concentrations of a substrate of 3A4, such as alprazolam (Xanax).
Inhibition. Certain drugs are not substrates for a particular enzyme but may nonetheless indirectly inhibit the enzyme and slow its metabolism of other drug substrates. For example, concurrent administration of a CYP 2D6 inhibitor, such as fluoxetine (Prozac), may inhibit the metabolism and thus raise the plasma concentrations of CYP 2D6 substrates, including amitriptyline (Elavil). If one CYP enzyme is inhibited, then its substrate accumulates until it is metabolized by an alternate CYP enzyme. Table 1-2 lists representative psychotropic drug substrates of human cytochromes P450 along with representative inhibitors.
Pharmacodynamics
The major pharmacodynamic considerations include molecular site of action; the dosage-response curve; the therapeutic index; and the development of tolerance, dependence, and withdrawal symptoms.
Molecular Site of Action
Psychotropic drugs may act at any of several molecular sites in brain cells. Some activate (agonists) or inactivate (antagonists) receptors for a specific neurotransmitter. Other drugs, particularly antidepressant drugs, bind to and block transporters that normally take up serotonin or norepinephrine from the synaptic cleft into the presynaptic nerve ending (reuptake inhibitors).
Some drugs block the passage of cations or anions through ion channels embedded in cellular membranes (channel inhibitors or blockers). Other drugs bind to and inhibit catabolic enzymes that normally inactivate neurotransmitters, prolonging the life span of the active neurotransmitters (e.g., monoamine oxidase inhibitors). Finally, several drugs have numerous molecular sites of action, although which sites are therapeutically relevant may remain unknown.
Dosage-Response Curves
The dosage-response curve plots the clinical response to the drug as a function of drug concentration (Figure 1-1). Potency refers to comparisons of the dosages of different drugs required to achieve a certain effect. For example, haloperidol (Haldol) is more potent than chlorpromazine (Thorazine) because about 2 mg of haloperidol is required to achieve the same therapeutic effect as 100 mg of chlorpromazine. However, haloperidol and chlorpromazine are equal in their clinical efficacy—that is, the maximum achievable clinical response.
Table 1-2 Representative Psychotropic Drug Substrates of Human Cytochromes P450 Along with Representative Inhibitors | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Therapeutic Index
The therapeutic index is a relative measure of a drug’s toxicity or safety. It is defined as the ratio of the median toxic dosage (TD50)—the dosage at which 50% of persons experience toxic effects—to the median effective dosage (ED50)—the dosage at which 50% of persons experience therapeutic effects. For example, haloperidol has a high therapeutic index, as evidenced by the wide range of dosages in which it is prescribed without monitoring of plasma concentrations. Conversely, lithium (Eskalith, Lithobid, Lithonate) has a low therapeutic index, thereby requiring the close monitoring of plasma concentrations to avoid toxicity.
Persons exhibit both interindividual and intraindividual variation in their responses to a specific drug. An individual may be hyporeactive, normally reactive, or hyperreactive to a particular drug. For example, whereas some persons require 50 mg a day of sertraline, other persons require 200 mg a day for control of their symptoms. An unpredictable, non–dosage-related drug response is called idiosyncratic. For example, diazepam (Valium) administered as a sedative paradoxically causes agitation in some persons.
Tolerance, Dependence, and Withdrawal Symptoms
A person who becomes less responsive to a particular drug over time is said to develop tolerance to the effects of the drug. The development of tolerance can be associated with the appearance of physical dependence, which is the necessity to continue administering the drug to prevent the appearance of withdrawal symptoms (also called discontinuation syndrome).
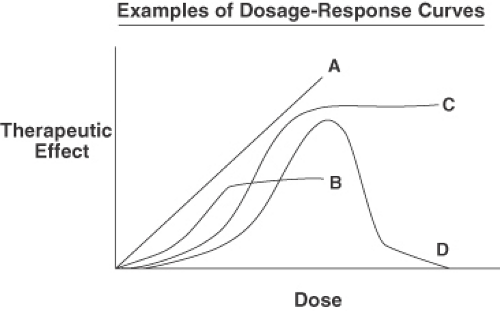 Figure 1-1. The dosage-response curves plot the therapeutic effect as a function of increasing dose, often calculated as the log of the dose. Drug A has a linear dosage response, drugs B and C have sigmoidal curves, and drug D has a curvilinear dosage-response curve. Although smaller doses of drug B are more potent than are equal doses of drug C, drug C has a higher maximum efficacy than does drug B. Drug D has a therapeutic window such that both low and high doses are less effective than midrange doses. |
Drug Interactions
Drug interactions may be either pharmacokinetic or pharmacodynamic, and they vary greatly in their potential to cause serious problems. Pharmacokinetic drug interactions concern the effects of drugs on their respective plasma concentrations, and pharmacodynamic drug interactions concern the effects of drugs on their respective receptor activities.
Pharmacodynamic drug–drug interactions causing additive biochemical changes may trigger toxic adverse effects. For example, monoamine oxidase inhibitors (MAOIs) when coadministered with either tricyclic antidepressants or selective serotonin reuptake inhibitors (SSRIs), may precipitate a serotonin syndrome in which serotonin is metabolized slowly and thus accumulates in excessive concentrations. The interaction of disulfiram (Antabuse) and alcohol is another example of toxicity caused by pharmacodynamic drug interactions.
Some clinically important drug interactions are well studied and well proven; other interactions are well documented but have only modest effects; and still other interactions are true but unproven, although reasonably plausible. Clinicians must
remember that (1) animal pharmacokinetic data are not always readily generalizable to humans; (2) in vitro data do not necessarily replicate the results obtained under in vivo conditions; (3) single-case reports can contain misleading information; and (4) studies of acute conditions should not be uncritically regarded as relevant to chronic, steady-state conditions.
remember that (1) animal pharmacokinetic data are not always readily generalizable to humans; (2) in vitro data do not necessarily replicate the results obtained under in vivo conditions; (3) single-case reports can contain misleading information; and (4) studies of acute conditions should not be uncritically regarded as relevant to chronic, steady-state conditions.
An additional consideration is one of phantom drug interactions. The person may be taking only drug A and then later receive both drug A and drug B. The clinician may then notice some effect and attribute it to the induction of metabolism. In fact, what may have occurred is that the person was more compliant at one point in the observation period than in another or there may have been some other effect of which the clinician was unaware. The clinical literature may contain reports of phantom drug interactions that are rare or nonexistent.
Informed clinicians need to keep these considerations in mind and to focus on the clinically important interactions, not on the ones that may be mild, unproven, or entirely phantom. At the same time, clinicians should maintain an open and receptive attitude toward the possibility of pharmacokinetic and pharmacodynamic drug interactions.
Drug Selection
There is no psychotropic drug that is effective in all patients with a given diagnosis. The ability of a drug to prove effective is only partially predictable and depends on the properties of the drug and the biology of the patient. Decisions about drug selection and use are made on a case-by-case basis, relying on the individual judgment of the physician. There are three factors in drug selection: (1) the drug, (2) the patient, and (3) the expertise and judgment of the prescribing physician. Each of these components affects the probability of a successful outcome. Table 1-3 outlines some factors influencing antipsychotic selection.
Therapeutic Indications
A therapeutic indication is a psychiatric diagnosis, as defined in the 10th revision of the International Statistical Classification of Diseases and Related Health Problems (ICD-10) or the fourth edition text revision of Diagnostic and Statistical Manual of Mental Disorders




Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
