(1)
Neurological Department, Municipal Hospital Dessau, Dessau-Rosslau, Germany
10.1 Introduction
The following chapter will focus on a couple of generalized or multifocal disorders affecting peripheral nerves, plexus and motor-neurons, which occasionally mimic focal neuropathies. Most of them are rather uncommon or rare. However, specialized knowledge will help to prevent patients undergoing surgical interventions or other therapies without benefit or even that they are harmed in a worst case scenario. Therefore, it is crucially important to always bear in mind the disorders described below and to perform – especially after failed surgery or unsuccessful treatment of a suspected focal neuropathy – an extensive differential-diagnostic approach. It would go beyond the scope of this book to go into every single detail. Instead, important facts are highlighted only so that physicians are helped to differentiate generalized respectively multifocal from focal neuropathies clinically. Furthermore the essential diagnostic pathways such as different imaging techniques and nerve conduction studies are explained.
10.2 Motor Neuron Disease
Behind the term “motor neuron disease” different degenerative disorders affecting the upper and/or lower motor neuron can be assumed. We will focus on amyotrophic lateral sclerosis (ALS) which was originally described by the French physician Jean-Martin Charcot [1]. The highest incidence of this rare disease is reported in Finland (2.4/100.000 subjects/year), [2]. Due to increased life spans of the population as well as improved diagnosis, the incidence is said to be increasing with a peak between the sixth and seventh decades [3, 4]. Most cases are sporadic, and approximately 10 % of them are said to be hereditary due to mutation of the SOD1 gene [5]. ALS affects both upper and lower motor neurons. Degeneration of upper motor neurons leads to a spastic paralysis, whereas degeneration of lower motor neurons results in muscle weakness and wasting [6]. Early manifestation of symptoms usually begins focally. Approximately two-thirds of cases start in the lower limbs [7]. This fact explains why some patients are initially misdiagnosed as cervical myelopathy (spasticity and hyperreflexia in their lower limbs in conjunction with muscle atrophy and weakness in the upper limbs), or as ulnar neuropathy or carpal tunnel syndrome (weakness and atrophy of intrinsic hand-muscles) as to be seen for instance in Fig. 10.1. In fact, 5 % of patients suffering from a motor neuron disease had cervical or lumbar decompression surgeries early in their course of complaints [8]. In the majority of cases, ALS rapidly progresses within 2–5 years after diagnosis, and death results from ventilatory weakness [9]. Currently, there exists no further curative treatment. The following three clinical signs may help to differentiate from a focal neuropathy in an early stage of ALS:
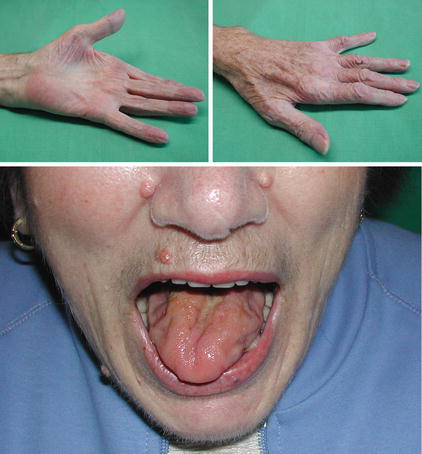
Fig. 10.1
Patient with amyotrophic lateral sclerosis. Note the focal beginning of the disease mimicking an ulnar neuropathy (upper part). Later on, most of the patients show signs of bulbar involvement (tongue atrophy)
No sensory symptoms (numbness, neuropathic complaints etc.) since primarily only the motor-neurons are affected
Widespread muscle twitching (fasciculations), even in clinically unaffected areas
Combination of muscle wasting with hyperreflexia (combination of signs of lower and upper motor-neuron)
These symptom-combinations should raise doubts regarding the eventual diagnosis of a focal peripheral neuropathy, and they should create the need for further examinations [10]. Typical signs of ALS are then moderately elevated levels of serum creatine kinase on one hand, and evidence of widespread denervation and reinnervation (in thoracic paraspinal muscles, muscles supplied by cranial nerves, etc.) in needle electromyography on the other. Sensory nerve conduction studies remain normal as well as motor nerve conduction velocity since the abnormality consists of axonal loss of motor fibers only [11, 12]. Signs of demyelination, especially abnormal temporal dispersion or conduction block, should instead raise serious doubts concerning diagnosis (see also multifocal motor neuropathy) [13]. Moreover, transcranial magnetic stimulation can show the involvement of the upper motor neuron [11].
Widespread fasciculations as well as muscle atrophies, additionally characterized by remodelling of muscle tissue with connective and fatty tissue, will lead to hyperechoic muscle appearance together with some decrease of muscle diameter in non-invasive high resolution ultrasound [14].
10.3 Diseases of Brachial and Lumbosacral Plexus
Among the different disorders of the brachial plexus there is one known as “neuralgic amyotrophy” that was first described in detail by Parsonage and Turner in 1957 [15]. We already mentioned this increasingly diagnosed disease in earlier sections, for example in Sect. 6.2.8. In addition to common idiopathic forms, a hereditary form is characterized by an autosomal-dominant trait. Recently, it has been reported that a mutation in the SEPT9 gene is detectable in several individuals suffering from such a hereditary neuralgic amyotrophy. The result is probably a myelin defect [16]. Etiology of the idiopathic form is unknown so far. Several mechanisms, especially of autoimmune origin, have been discussed [17]. Compared to motor neuron diseases, the incidence of 2–3/100.000/ individuals/year regarding the idiopathic form is rather low [18]. The typical clinical course, sometimes with attacks, consists of three stages [19]:
Stage one is characterized by a sudden onset of severe aching or throbbing pain reaching a score between 8 and 9 on a pain-scale ranging from 1 to 10. Typically the pain arises from the cervical spine, shoulder or scapular region and radiating down to the arm. This stage usually lasts for 4 weeks on average.
In stage two mostly a motor deficit with a “patchy” distribution pattern in the territory of one or different peripheral nerves or nerve roots occurs, characterized by weakness, paralysis and muscle atrophy, outlasting the pain for a long time. In the majority of cases (71 %), the upper and middle brachial plexus with or without long thoracic (winged scapula) or suprascapular nerve are involved but nearly every nerve in the brachial plexus can be affected. Sensory symptoms are less clinical prominent.
In the third stage – lasting 3 years or more – one-third of the patients experience a complete recovery, whereas in two- thirds the overall recovery is less favourable with persisting pain and residual paresis [19].
Attacks are more relapsing in patients with hereditary neuralgic amyotrophy (74.5 %) than with the idiopathic form (26.1 %). Antecedent events were monitored in approximately half of the cases, primarily infection, surgery and exercise [19].
In a smaller subgroup of patients neuralgic amyotrophy may predominantly involve the anterior interosseus nerve (see Sect. 8.13.1 and Fig. 6.24) or the radial nerve (see Sects. 8.9 and 8.10.1) as well as the lower brachial plexus (Fig. 6.25). This applies for women especially [19]. Here, the typical clinical course of attack-like onset helps to differentiate neuralgic amyotrophy from a true focal compressive neuropathy.
An important differential diagnosis of a painful brachial plexus lesion is infiltration by a malignant tumor/metastasis. This fact should always be borne in mind, particularly concerning patients with history of lung or breast cancer.
Laboratory tests and lumbar puncture often reveal things as normal and may help to exclude other conditions with similar symptoms (e.g. neuroborreliosis) [19]. Approximately 3 weeks after onset needle electromyography can demonstrate the extent and distribution of the axonal damage in 96 % of cases. In particular, subtle denervation in muscles not supplied by a single nerve as well as in muscles of the contralateral side may help to differentiate from a focal neuropathy [17]. In 6 % of cases, by means of MR imaging, a T2 lesion or hypertrophy of the brachial plexus was described [19]. There are no actual reports about ultrasound imaging regarding the neuralgic amyotrophy. In our own experience, hypoechoic and long-segmental CSA enlargement of the affected roots, of parts of the brachial plexus, of peripheral nerves, or of even single fascicles can be detected weeks after onset of the disease but not at the beginning (Figs. 6.11, 6.24, 6.25, and 8.19). Pham et al. [20] found 20 cases of neuralgic amyotrophy mimicking an interosseus anterior syndrome a T2-lesion of the corresponding fascicle group within the median nerve at upper arm. It remains unclear at the moment whether these changes will be reversible. Perhaps there is also an affiliation with spontaneous torsional nerve neuropathy in some cases, as demonstrated in Fig. 8.11 [21]. Although neuralgic amyotrophy is a self-limiting condition, some supportive therapies may be helpful. Most important is adequate analgesia during the initial phase of the disease as well as physical therapy. Some very preliminary data on patients treated with corticosteroids suggest that the course of the symptoms might thus be favourably influenced [19].
10.4 Hereditary Neuropathies (CMT I, HNPP)
10.4.1 CMT – Charcot-Marie-Tooth Disease
CMT – also known under the term hereditary motor-sensory neuropathy (HMSN) – was first described by Charcot, Marie and Tooth in 1886. It affects approximately 1 in 2,500 individuals [22, 23]. It is represented by a heterogeneous group of different hereditary disorders whereby CMT Types Ia and IIa are to be found most frequently [24]. CMT Type Ia is associated with a duplication on chromosome 17 which contains the gene encoding peripheral myelin protein 22kD (PMP22) leading to a demyelinating neuropathy [25]. A mutation in the gene encoding kinesin, a protein that is involved in microtubule-mediated axonal transport, leads to CMT Type IIb, clinically represented by an axonal neuropathy [26]. Both CMT I and II are usually inherited as an autosomal dominant trait. Regarding further, mostly rare, types and subtypes please refer to specialist literature [27]. The classical phenotype of CMT affects individuals younger than 20 years of age and progresses slowly (Fig. 10.2). Muscle weakness/atrophy and sensory dysfunction usually starts distal-symmetric at the lower extremities, characterized by a foot drop/peroneal atrophy with steppage gait and a sock-shaped sensory impairment [28, 29]. In an advanced stage of the disease, distal and symmetric muscle weakness and atrophy of the intrinsic hand muscles (Fig. 10.3) as well as the not always glove-shaped sensory impairment may be misdiagnosed as carpal tunnel syndrome or ulnar neuropathy at the elbow. Moreover, skeletal abnormalities such as pes cavus, hammer toes and scoliosis are very characteristic [28, 29]. Beyond this classical phenotype, some variants without risk of confusion with a focal neuropathy have been described: infantile and early onset forms are characterized by a mostly severe muscle weakness (“floppy infant”), severe sensory impairment, and they may end fatally [27].
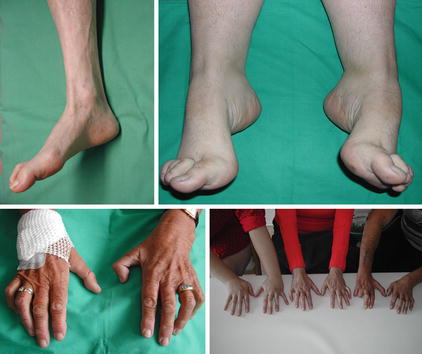
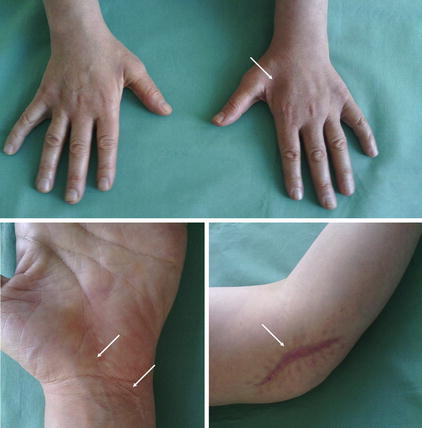
Fig. 10.2
Clinical presentation of hereditary motor-sensory neuropathy (HMSN). Upper part: Pes cavus and hammer toes. Lower part: HMSN mimicking an ulnar neuropathy. Right image shows an HMSN family (daughter, mother and brother of the mother from left to right)
Fig. 10.3
HMSN mimicking multiple entrapment syndromes (carpal tunnel syndrome, Guyon’s canal syndrome as well as ulnar neuropathy at the elbow). After surgical decompression at the left site (arrows) a worsening of the symptoms appeared
Suspected diagnosis is established by the typical clinical signs mentioned above, a positive family history, electrodiagnostic testing and high resolution ultrasound. As stated in Chap. 5, sensory and motor nerve conduction studies are the primary tests to distinguish between axonal and demyelinating neuropathies. CMT Type I (demyelinating) is characterized by a slowing of motor nerve conduction velocities ~20 m/s and prolonged distal motor latencies [25] (Fig. 10.4). Except for in CMT I, such slow conduction velocities regularly only occur in some neuropathies associated with a gammopathy. Moreover, in contrast to acquired neuropathies, the slowing of nerve conduction velocities in an inherited neuropathy occurs uniformly, whereas acquired neuropathies usually exhibit asymmetric or non-uniform slowing [30]. However, the axonal loss in CMT Type II results in a reduction of the amplitude of CMAP or SNAP.
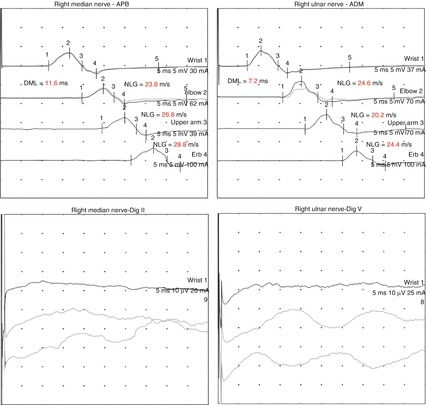
Fig. 10.4
Corresponding EDX. Note the uniform slowing of motor nerve conduction velocities as well as the prolonged distal motor latencies in the ulnar and median nerve suggesting a generalized demyelinating process. Sensory nerve action potentials could not be obtained. The same results revealed nerve conduction studies at the opposite site (not shown). At the lower extremities (not shown) both compound muscle action potentials and sensory nerve action potentials were absent
The most recent diagnostic tool is the high resolution ultrasound. Individuals with CMT have a profound (CMT I) or modest (CMT II) enlargement of peripheral nerve cross-sectional area [31] (Fig. 10.5). If the evidence points to CMT disease, one of the most important issues consist of genetic testing and counselling to confirm the diagnosis [32]. There exists no curative treatment at the moment but supportive therapies may gain a partly symptom control [27]. In summary, important clues for a CMT are:
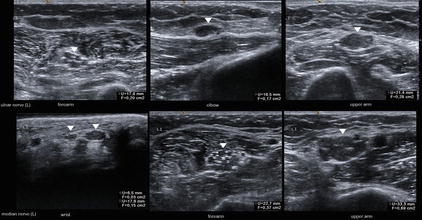
Fig. 10.5
Corresponding ultrasound image of the left ulnar and median nerve (arrowheads) demonstrates a severe hypoechoic swelling of fascicles and the entire nerve far away from the typical sites of entrapment
Clinical signs of a symmetric “focal” neuropathy such as both-sided carpal tunnel syndrome and/or ulnar neuropathy combined with signs of a distal-symmetric sensory-motor neuropathy of the legs in individuals of relatively young age
Pes cavus, hammer toes
Uniformly slowed motor nerve conduction velocities ~20 m/s, prolonged distal motor latencies (CMT I), signs of axonal loss (CMT II)
Profound (CMT I) or modest (CMT II) enlargement of peripheral nerve cross-sectional area on ultrasound
Positive family history
10.4.2 HNPP – Hereditary Neuropathy with Liability to Pressure Palsies
Another important hereditary neuropathy mimicking a focal neuropathy is the HNPP, a disorder that was probably first described by De Long in 1947 [33]. The disease is also inherited as an autosomal dominant trait. In contrast to CMT I it is caused by a deletion on chromosome 17, containing the gene encoding the peripheral myelin protein 22kD (PMP22) [34]. The abnormal myelin may be seen in histo-pathological studies in the form of a focal, sausage-shaped thickening of myelin sheaths, also called tomacula [35]. Typically, the disease has an early onset, and it likely occurs in adolescents and in young adults [35]. Due to the abnormal myelin protein, affected individuals suffer from multiple remitting and relapsing nerve entrapments. Just slight pressure, which nerves easily tolerate normally, may induce a nerve impairment in subjects with HNPP (Fig. 10.6). Nerve entrapment often occurs in the peroneal, ulnar and radial nerves, but it has also been described in the brachial plexus [36, 37]. The clinical manifestations include both sensory impairment and muscle weakness.
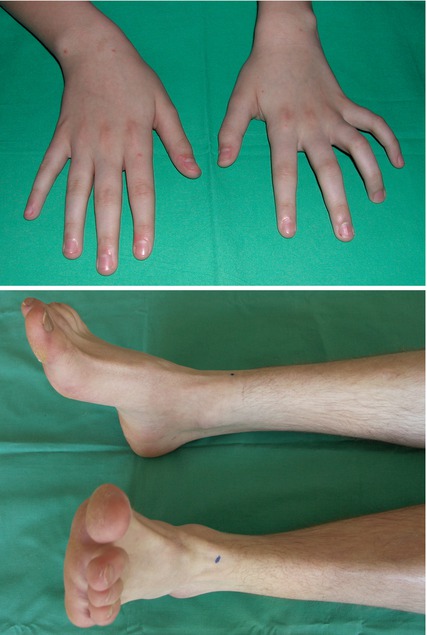
Fig. 10.6
Clinical presentation of HNPP in a 13-year-old boy. Remitting and relapsing nerve entrapments of the left ulnar nerve after excessive computer gaming (above). After 10 weeks the patient developed a palsy of the right peroneal nerve after leg crossing (below)
Electrophysiological studies typically demonstrate features of demyelination such as prolonged distal motor latencies, ubiquitous reduced sensory and motor nerve conduction velocities and a segmental slowing of nerve conduction across the typical entrapment sites. However, nerve conduction is not as low as in CMT I [38]. These changes may generally be observed in asymptomatic individuals at asymptomatic entrapment sites [39].
As in CMT, high resolution ultrasound of the peripheral nerves provides a novel tool in the diagnosis of HNPP. The average nerve CSA was greater in CMT1 than in HNPP and the most common site of nerve hypertrophy was at sites of entrapment. It is again associated with profound enlargement of peripheral nerve cross-sectional area [31]. HNPP has a favourable prognosis because of possibly spontaneous recovery within weeks or months. In some case reports it has been stated that patients also have received a benefit from surgery of the entrapped nerves [40]. However, it remains unclear whether it was due to surgery or to the natural course of the disease.
Today, it is easy to confirm the diagnosis by a genetic test, whereas meanwhile a biopsy of the sural nerve is unnecessary [41]. In summary, important clues for HNPP are:
Remitting and relapsing nerve entrapment in adolescents and young adults after just slight pressure, mostly seen in the peroneal, ulnar and radial nerves with a sensory-motor impairment and spontaneous recovery
Ubiquitous slight slowing of motor and sensory nerve conduction velocities, prolonged distal motor latencies, segmental slowing of nerve conduction across the typical entrapment sites, and changes in asymptomatic individuals
Profound enlargement of peripheral nerve cross-sectional area on ultrasound most frequently on entrapment sites
Positive family history
10.5 Autoimmune Neuropathies
10.5.1 Chronic Inflammatory Demyelinating Neuropathy and Variants
Among the autoimmune-induced neuropathies, we have to take into account some conditions which again may be confused with a focal neuropathy. Most of them are mainly characterized by a chronic course. The classical prototype is a disease also referred to as chronic inflammatory demyelinating neuropathy (CIDP). Its etiology is completely unknown to date. However, similarity to the Guillain-Barre syndrome, as well as response to an immune-modulating therapy, suggest an autoimmune origin [42]. The primary hallmark of CIDP is demyelination followed by secondary axonal loss [43]. The prevalence has been reported to be 1–8 affected individuals within a population of 100,000 inhabitants [44]. These are likely underestimates, since the condition is not always easy to diagnose. Therefore it is supposed that a considerable percentage of the affected subjects are overlooked or misdiagnosed [45]. CIDP is characterized by predominant motor symptoms persisting for more than 2 months together with reduced or absent tendon reflexes. Either progressive or relapsing-remitting weakness may start asymmetrically at the onset of the disease, but it typically affects symmetrically the distal and/or proximal muscles of the extremities, whereas involvement of the cranial nerves is uncommon. In addition, sensory loss such as numbness may also be present. Some of the affected individuals show a self-limiting course [42]. In contrast to hereditary neuropathies, family history is negative. Besides the classical CIDP, there are conditions with an atypical clinical presentation (see below), some of which may be considered variants and others distinct disorders. Due to this heterogeneity as well as the lack of a specific diagnostic test, several diagnostic criteria have been suggested. They all have a high specificity but are less sensitive [46–48]. For instance, the frequently used criteria of the American Academy of Neurology (evidence of demyelination in nerve conduction studies, elevated CSF protein level and signs of demyelination on nerve biopsy) miss more than 50 % of the patients suffering from CIDP, but they approach a specificity of 100 % [49]. Therefore, it has been suggested to start with an immune-modulating therapy if the typical clinical symptoms mentioned above are present and if additional criteria support the diagnosis [42]. Nerve conduction studies play a key role since they since they demonstrate the underlying process of demyelination in various nerve segments by detection of slowed nerve conduction velocities, prolonged distal motor and F-wave latencies as well as conduction block or abnormal temporal dispersion (please also refer to Chap. 5). Electrophysiologic criteria of the American Academy of Neurology require at least three demyelinating range abnormalities in two nerves; however, they are less sensitive as stated above [46]. Moreover, in 90 % of the affected individuals, an elevated liquor protein is present, and in 29 % of patients antibodies to P0 protein were identified [50]. Furthermore, sural nerve biopsy may yield evidence of demyelination, but it is considered to be less sensitive and less specific [42].
Recently, imaging modalities have been introduced in the diagnosis of CIDP. Hypertrophy of the lumbar or cervical nerve roots as well as of the brachial or lumbosacral plexus and/or gadolinium uptake in MRI may support the diagnosis (Fig. 6.23) [51, 52]. Additionally, high resolution ultrasound provides a cost-effective tool for the detection of focal, multifocal or generalized alterations of the peripheral nerves and plexus. Ultrasonographic findings that have been reported in CIDP cases are characterized by an modest enlargement of cervical nerve roots and non, focal or multifocal peripheral nerve enlargement (see Sect. 6.1.8 concerning HRUS and Sect. 6.2.8 concerning MRI) [53]. However, in some cases focal enlargement corresponding to sites of conduction block was demonstrated. Similar findings are reported to occur in multifocal motor neuropathy (see Sect. 10.5.2), but further investigations are required to determine the potential diagnostic value of these methods [31, 54]. Treatment is immune-modulation either with corticosteroids or alternatively with intravenous immune globulin [55, 56]. As in CMT I, chronic inflammatory demyelinating neuropathy may be confused with multiple entrapment syndromes of the upper extremities such as both-sided carpal tunnel syndrome due to prolonged distal motor latencies. Therefore, doubts concerning the diagnosis of a focal neuropathy should arise if:
There are clinical signs, or there is electrodiagnostic evidence, of a demyelinating lesion concerning multiple nerves, especially in the proximal parts of the upper extremity (proximal conduction block)Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree







