Genetics of Epilepsy Syndromes
Ortrud K. Steinlein
Introduction
The term epilepsy describes a heterogeneous group of disorders, with a lifetime cumulative incidence of 3%. There are disorders in which seizures are the main or only symptom, and those in which seizures are just one symptom among others. The reasons why individuals develop epilepsy are numerous. Examples for mostly nongenetic causes of epilepsy are trauma, tumor, and infections. On the other end of the spectrum are epilepsies that have no obvious etiology but are suspected to be mainly genetic in origin. The latter group of epilepsies has been termed idiopathic, in the meaning that they are “not preceded or occasioned by another disorder”32 and that “there is no underlying cause other than a possible inherited predisposition” and “a symptomatic origin is neither detected nor suspected.”33 By now the distinction between symptomatic and idiopathic epilepsies has become blurred because of our growing knowledge about the complex and diverse etiology of epilepsies. Genes for several idiopathic epilepsies have already been discovered, and most of these genes are able to (at least temporarily) change the functional state of the brain. Some of them are expressed during embryogenesis, rendering it possible that some of them might even interfere with normal brain development and cause subtle changes of the brain’s microanatomy. On the other hand, there is a large group of epilepsies and disorders with epilepsy that have been termed “symptomatic” because they are due to known metabolic, neurodegenerative, or structural brain damage. Many of these disorders have by now shown to be caused by clearly defined genetic factors. It is likely that in the future the terms nongenetic epilepsies, genetic epilepsies, and genetic disorders with epilepsy will more and more replace terms like idiopathic and symptomatic. For this chapter exemplary members of both groups have been chosen to illustrate principal etiologic categories of the disorder “epilepsy” and trace the various genetic pathways to epileptogenesis.
Inheritance and Genes in Common Idiopathic Epilepsies
About 30% to 40% of all epilepsies, especially during childhood and adolescence, are summarized under the term idiopathic epilepsies. They can be roughly divided into the group of common, mostly generalized idiopathic epilepsies (IGEs) and the various rare monogenic forms of partial or generalized idiopathic epilepsy. The first group, IGE, includes age-related subtypes like juvenile myoclonic epilepsy, childhood absence epilepsy, juvenile absence epilepsy, and grand mal epilepsy on awakening. The high concordance rates found in twin studies support the hypothesis of an almost complete genetic etiology of IGE.10 Nevertheless, recurrence risks in first-degree relatives of patients with IGE are considerably lower than in monogenic disorders, arguing for an oligogenic or polygenic model of inheritance. It can be assumed that several susceptibility genes are involved in each patient, and that the interaction between these gene loci is multiplicative rather than additive. Some IGE genes might determine the seizure threshold by influencing neuronal excitability, while other susceptibility genes might be responsible for the age of onset and therefore the seizure subtype.146
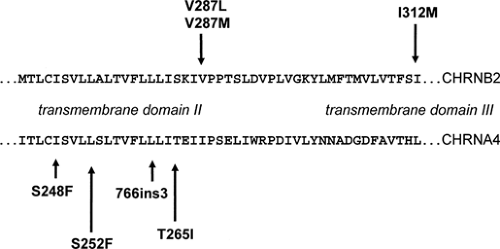 FIGURE 1. Mutational spectrum in autosomal dominant nocturnal frontal lobe epilepsy. The parts of nicotinic acetylcholine receptor subunit genes CHRNA4 and CHRNB2 containing transmembrane domains II and III are shown. |
Two different models have been proposed to explain the complexity of genetic factors and gene–gene interactions in common IGE. In the ancestral common variant complex epilepsy model (ACVCE), the existence of common IGE-associated alleles is assumed. In homogeneous patient samples, such alleles should be detectable by association studies. The second model, named multiple rare variant complex epilepsy model (MRVCE), describes the existence of many different rare, slightly deleterious mutations that cannot be detected by association studies. Both models would be able to explain the complex inheritance patterns seen in IGE, and both common and rare gene variants have already been identified.86 For example, both the ACVCE and the MRVCE models of complex inheritance apply to sequence variants in the T-type calcium channel gene CACNA1H on chromosome 16p13.3. Several rare CACNA1H variants that markedly alter channel properties have been found in patients with childhood absence epilepsy (F161L, E282K, V831M) or other IGE subtypes (P618L, G755D). Interestingly, a common CACNA1H sequence variant exists that, in combination with one of the above-mentioned rare variants, is able to alter the calcium channels’ biopharmacologic properties in a way that neither the rare nor the common variant separately do.67 Additional examples consistent with both the ACVCE and the MRVCE models are found in the GABRD gene that codes for the δ-subunit of the γ-aminobutyric acid (GABA)A receptor. The GABRD sequence variant E177A was described in one family and, compared to wild-type receptors, was shown to significantly reduce the GABAA receptor’s maximal current. A more common GABRD variant, R220H, was detected in both epilepsy patients and controls. Receptors heterozygous for R220H had a significantly decreased peak current in comparison with the wild type; thus, R220H may act additively as a susceptibility factor in combination with other, yet to be identified sequence variants.38 Nevertheless, CACNA1H, GABRD, and other genes that are presently discussed as susceptibility factors probably still account only for a very small fraction of the genetic contribution to common IGE syndromes. Many more genes remain to be discovered, a process that has so far been most successful in the monogenic forms of idiopathic epilepsy described in the next paragraphs.
Idiopathic Epilepsies Are Often Channelopathies
The progress in identification of genes in monogenic forms of epilepsy illustrates the important role of ion channels in epileptogenesis. A great variety of different ion channels regulate brain excitability and prevent hypersynchronization of neuronal networks. Mutations in any of those ion channels might change the delicate balance between excitatory and inhibitory input, resulting in recurrent firing, hyperexcitability, and, finally, seizures.
Familial Nocturnal Frontal Lobe Epilepsy
In 1994, autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) was first described as an inherited form of partial epilepsy. ADNFLE is characterized by clusters of brief motor seizures, which occur mostly during non–rapid eye movement (REM) sleep.106 Nonspecific aura phenomena including epigastric, sensory, or psychic symptoms sometimes precede the seizures. The seizures start with gasps, grunts, or some vocalizations followed by thrashing hyperkinetic activity or tonic stiffening of the limbs, often with superimposed clonic jerking. The consciousness is only infrequently impaired. Seizure frequency can be highly variable even within the same family. In some of the patients seizures are frequent and tend to cluster, while in others seizure occurrence is more sporadic and long periods of spontaneous remission are reported. In general, seizure frequency tends to decrease in later adulthood. Physical examinations and brain imaging results are usually normal. Interictal electroencephalographic (EEG) abnormalities are rarely seen, and only some patients show ictal epileptiform activity. The onset of ADNFLE is usually during childhood or early adolescence, with a penetrance of 70% to 80%. Seizures are often, but not in all patients readily controlled by antiepileptic drugs, especially carbamazepine.
Mutations in two genes have so far been identified in different ADNFLE families.46,121 CHRNA4 and CHRNB2 encode the α4- and β2-subunits of the neuronal nicotinic acetylcholine receptor (nAChR), respectively. The first CHRNA4 mutation, a S248F amino acid exchange within the second transmembrane domain, was identified in 1995.121 Descriptions of other nAChR mutations, either in CHRNA4 or in CHRNB2, followed.62,73,98,122 With the exception of one mutation (CHRNB2-I312M in transmembrane domain III), all known ADNFLE mutations are located within the second transmembrane domain. Both the second and the third transmembrane domain contribute to the walls of the ion channel; thus, it seems that only mutations that have a direct effect on the ion pore are able to cause ADNFLE. In total, four CHRNA4 and three CHRNB2 mutations have been described. These include six amino acid exchanges and one small in frame insertion. Some of those mutations have been found again in unrelated families. They offer the opportunity to study the effects differences in genetic and ethical backgrounds might have on the penetrance of the disorder and the clinical features associated with these mutations (Fig. 1).
The nAChRs, like the glycine, GABAA, and serotonin receptors, are members of the large family of ligand-gated ion channels. They are constituted by the assembly of five subunits arranged pseudosymmetrically around the central axis, forming a cation-selective ion pore. Pharmacologic and ligand-binding studies have shown that, depending on their subunit composition, the different subtypes of homo- or heteropentameric nAChRs not only vary with respect to their expression patterns in the brain, but also display different biopharmacologic channel properties.11 Both α4- and β2-subunits are found in almost all brain regions, and the heteropentameric α4/β2 receptor is thought to be the major high-affinity nicotine nAChR subtype in mammalian brain. The surprising and so far not very well understood part is that an nAChR subtype ubiquitously present in brain is able to cause a partial type of epilepsy rather than a generalized one. In support of the recognized importance of the cholinergic system in brain functions, recent work has shown that nAChR mutations not only cause epilepsy, but can also be associated with additional neurologic and psychiatric features or cognitive deficits.29,62,80 Examples are the CHRNA4-776ins3 mutation and the CHRNA4-S252L mutation. The 776ins3 mutation was found in 11 members of a large Norwegian ADNFLE family. Six of the 11 mutation carriers not only had epilepsy, but also had serious psychiatric problems, mostly schizophrenia, negative symptoms of schizophrenia, or recurrent unclassified psychosis. These observations suggest that the 776ins3 mutation may be a risk factor for psychiatric disorders.80 The S252L mutation has been described in two unrelated families of Korean and Japanese origin. In both families the seizures not only tended to be resistant to carbamazepine treatment, but were also associated with a high rate of mental retardation and/or behavioral problems.29,62 The similarities of the clinical phenotype in two families with different genetic background but the same S252L-ADNFLE mutation strongly suggest that the associated neurologic features are mutation specific rather than caused by modifying genes or environmental factors (Fig. 2).
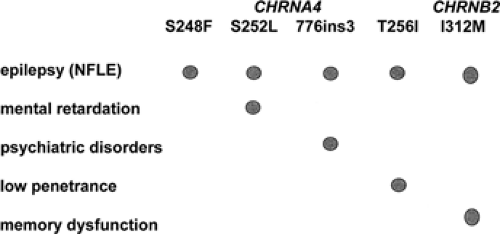 FIGURE 2. Clinical features frequently associated with different nicotinic acetylcholine receptor mutations in autosomal dominant nocturnal frontal lobe epilepsy families. NFLE, nocturnal frontal lobe epilepsy. |
Functional analysis carried out with recombinant nAChR receptors in Xenopus oocytes or HEK cells, including patch clamp characterization of several known ADNFLE mutations, revealed different biopharmacologic profiles.11 Co-expression of the S248F mutation and the wild-type CHRNA4 allele yielded acetylcholine-evoked currents of amplitudes comparable to wild-type receptors but with a higher sensitivity for the natural agonist. Receptors carrying either the 776ins3, S252L, or V287M mutation displayed increased acetylcholine sensitivity but to different degrees compared with the S248F mutation. A reduction of calcium permeability was observed for the mutants S248F and 776ins3 but not for the S252L mutation.11 Increased acetylcholine sensitivity and therefore a gain-of-function effect is the only functional feature common to all known ADNFLE mutations. It is tempting to speculate that the particular functional signatures of each mutant contribute to the above-described observations of associated neurologic features in ADNFLE, while the gain-of-function effect might be responsible for the epilepsy phenotype. The latter part of this hypothesis is supported by the observation that, compared to wild-type litter mates, mice carrying gain-of-function mutations in the CHRNA4 gene are dramatically more sensitive to nicotine-induced seizures. Furthermore, nicotine application in these mice resulted in enhanced hippocampal theta rhythms, as demonstrated by in vivo electrophysiologic recordings. This observation would be consistent with a model in which gain-of-function effects in presynaptically located nAChRs activate inhibitory GABAergic interneurons in the neocortex and hippocampus. The enhanced GABA release could then increase network excitability by inhibiting inhibitory pathways. Alternative models for the epileptogenic effect of nAChR mutations could include a disturbance of hippocampal “peacemaker” theta activity due to increased input from septal cholinergic neurons.
Benign Familial Neonatal Convulsions
Benign familial neonatal convulsions (BFNC) is a rare autosomal dominant seizure disorder of the newborn (see also Chapter 223). The disorder is characterized by an age of onset between the first day and, latest, the fourth month of life. The seizures are mostly unprovoked, generalized, or multifocal and of the tonic and/or clonic type. The course of the disorder is often benign and self-limiting, and, with or without pharmacotherapy, in most patients the seizures remit spontaneously within a few days or weeks.103,137 Later in life seizures can reoccur in up to 15% of the patients, starting mostly at school age or in young adulthood. These so-called late-onset seizures tend to be infrequent and are often provoked, for example, by lack of sleep. Several untypical BFNC patients with a more severe course of the disorder have been described. These patients often show a higher frequency of seizures and seizures still occur after the age of 4 months. Follow-up studies often revealed moderate delays of psychomotor development, and in some patients have a family history positive for epilepsy and mental retardation. Two BFNC families have come to attention in which a mutation carrier developed drug-resistant seizures and/or epileptic encephalopathy shortly after birth, resulting in severe psychomotor retardation. In one of the two severely affected index patients a de novo mutation was found, while the second index patient inherited his mutation from a parent with typical “benign” BFNC.36,109 It remains unclear if these two patients had a second yet unrecognized condition or if certain risk factors (e.g., perinatal hypoxia) in combination with a BFNC mutation increase the risk for such an unfavorable outcome.
The molecular basis of BFNC is a mutation in the voltage-gated potassium channel genes KCNQ2 or KCNQ3 (chromosome 20q13.3 and 8q24, respectively).13,27,115 Both the KCNQ2 and KCNQ3 genes encode ion channel subunits that are composed of six transmembrane domains. The subunits have identical structures including a voltage sensor in transmembrane domain 4, a loop between transmembrane domains 5 and 6 that builds the ion channel pore, and a long C-terminal region of unknown function. The BFNC mutations found so far in KCNQ2 are either missense mutations located in one of the transmembrane domains or truncating mutations (including nonsense, insertion/deletions, and splice site mutations) located mostly in the C-terminal region. Most mutations are private, meaning that they have not been found in other BFNC families. So far, only three mutations have been found in KCNQ3. All known KCNQ3 mutations are missense mutations located within the vicinity of the pore region.
KCNQ2 and KCNQ3 encode subunits of the M-channel, a very slowly opening and closing potassium channel that is ubiquitously found in brain.141 The M-current, first discovered some 20 years ago,21 is a powerful controller of neuronal repetitive firing. M-currents regulate the number of action potentials of individual neurons by opposing sustained membrane depolarization. M-channels activate at membrane potentials that are more negative than the action potential threshold and at which few other ion channels are active. Therefore, they can be assumed to have a pivotal role in the stabilization of membrane potentials and are likely to control excess neuronal excitability and prevent seizures. Expression studies in Xenopus oocytes and HEK cells have shown that BFNC mutations cause only modest reductions (20% to 30%) of potassium currents in reconstitution experiments. It seems, therefore, that even slight alterations of M-channel activity are sufficient to cause seizures.13
Exceptions to this haploinsufficiency concept are some rare KCNQ2 mutations that appear to have a dominant negative effect on channel function, as demonstrated by a >50% reduction in current magnitude. One of these mutations is KCNQ2/R207W, which causes the BFNC/myokymia syndrome. The mutation abolishes the third of six positive charges in transmembrane domain 4 that is thought to represent the voltage sensor in the cation channel superfamily. The R207W amino acid exchange was the first KCNQ2 mutation described that causes a disorder with symptoms outside the central nervous system; it is associated not only with episodic symptoms, but also with continuous symptoms. On the clinical level, heterozygosity for the R207W mutation causes both BFNC and myokymia, a spontaneous and repetitive involuntary contraction of muscle fiber groups.37 In the BFNC/myokymia syndrome, multifocal or generalized tonic–clonic convulsions typically start around day 3 after birth and disappear spontaneously after a few weeks or months. Electromyographic (EMG) recordings in the patients are initially normal but,
starting in infancy, reveal spontaneous repetitively discharged normal motor unit potentials of 20 to 60 msec in duration. Generalized myokymia with spontaneous finger twitching occurs some years later. The association with both central and peripheral neurologic symptoms might be explained by the unusual electrophysiologic profile of the underlying mutation. R207W causes a loss of K+ current, whose magnitude depends strongly on the pattern and time course of depolarization. With short periods of depolarization the loss of current is more severe than with other known BFNC mutations. The dominant negative effect is not observed with longer periods of activation. Thus, at long (>1 second) trains of action potentials or during seizure activity, R207W mutant channels may be activated significantly and may even reach wild-type levels. It is possible that the time-dependent dominant negative effect of R207W establishes itself only in the peripheral nervous system and that this might be the reason for peripheral neurologic symptoms like myokymia. This hypothesis rests on the assumption that the neuronal activity of motoneurons is interrupted by longer quiescent periods than is the activity of central neurons whose hyperexcitability triggers seizures. Under such conditions, the dominant negative effect of the R207W mutation would have consequences for motoneurons but not for central neurons.37
starting in infancy, reveal spontaneous repetitively discharged normal motor unit potentials of 20 to 60 msec in duration. Generalized myokymia with spontaneous finger twitching occurs some years later. The association with both central and peripheral neurologic symptoms might be explained by the unusual electrophysiologic profile of the underlying mutation. R207W causes a loss of K+ current, whose magnitude depends strongly on the pattern and time course of depolarization. With short periods of depolarization the loss of current is more severe than with other known BFNC mutations. The dominant negative effect is not observed with longer periods of activation. Thus, at long (>1 second) trains of action potentials or during seizure activity, R207W mutant channels may be activated significantly and may even reach wild-type levels. It is possible that the time-dependent dominant negative effect of R207W establishes itself only in the peripheral nervous system and that this might be the reason for peripheral neurologic symptoms like myokymia. This hypothesis rests on the assumption that the neuronal activity of motoneurons is interrupted by longer quiescent periods than is the activity of central neurons whose hyperexcitability triggers seizures. Under such conditions, the dominant negative effect of the R207W mutation would have consequences for motoneurons but not for central neurons.37
In mice, KCNQ2 knockout experiments resulted in early postnatal lethality in homozygous animals, while hemizygous knockout mice developed and behaved normal but showed an increased sensitivity to pentylenetetrazole, an epileptic inducer.142 Transgenic mice that conditionally express a dominant negative KCNQ2 pore mutation in the brain only during defined developmental periods demonstrated the critical role of M-channel activity in early postnatal brain development. Suppression of the M-current during the first postnatal weeks was associated with spontaneous seizures, behavioral hyperactivity, and morphologic changes in the hippocampus.96 These results support the notion that M-currents are critical determinants of cellular and neuronal network excitability. The changes in brain morphology found in transgenic mice also raise the interesting question of whether BFNC caused by KCNQ2 mutations in humans might be (at least in part) due to submicroscopic structural brain changes (morphologic etiology) rather than exclusively to a reduction of M-currents (functional etiology).
Benign Familial Infantile Convulsions
Benign familial infantile convulsions (BFIC) can be distinguished from BFNCs by a later age at onset135 (Fig. 3). The autosomal dominant disorder is characterized by the onset of seizures around 6 months of age (range 3 to 9 months). Seizures occur in clusters and respond well to antiepileptic drug treatment. The seizures are partial with secondary generalization and in most patients spontaneous remission has occurred by the age of 12 months. Usually no subsequent neurologic abnormalities develop. Two putative loci were reported, BFIC1 (19q) and BFIC2 (16p12-q12), but no genes have been identified yet.23,54 Interestingly, three other disorders with overlapping neurologic features map to the same region as BFIC2: The infantile convulsions and choreoathetosis syndrome (ICCA; OMIM 602066),124 paroxysmal kinesigenic choreoathetosis (PKC; OMIM 128200),129 and a syndrome comprising rolandic epilepsy, paroxysmal exercise-induced dystonia, and writer’s cramp (EPRPDC; OMIM 608105).53 Therefore, the possibility exists that the four disorders are allelic.
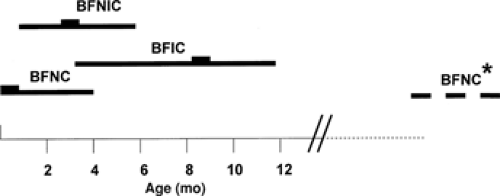 FIGURE 3. Age of onset and remission in benign familial neonatal convulsions (BFNC), benign familial neonatal/infantile convulsions (BFNIC), and benign familial infantile convulsions (BFIC). |
Febrile Seizures
Febrile seizures (FSs) affect 5% to 10% of children under the age of 6 years. Twin and family studies demonstrated the involvement of genetic factors in the etiology of febrile seizures. In most patients an oligo- or polygenic background rather than a monogenic one can be assumed, and a substantial degree of heterogeneity is likely. However, rare families with an apparent autosomal dominant mode of inheritance have been described, and linkage studies identified several putative gene loci, including FEB1 on chromosome 8q13-q21, FEB2 on 19p, FEB3 on 2q23-q24, FEB4 on 5q14-q15, FEB5 on 6q22-q24, and FEB6 on 18p11.2.64,81,88,90,92,138 In one family with febrile and afebrile seizures, a nonsense mutation (S2652X) was identified in the MASS1 gene. MASS1 is part of the large G-protein coupled receptor gene VLGR1.90 The mutation causes a deletion of the C-terminal 126 amino acid residues in the predicted MASS1 protein. If confirmed in independent families, MASS1 can be regarded as a rare cause for familial febrile convulsions. In another family a missense mutation in the SCN1A gene chromosome 2q23-24 has been found to cosegregate febrile seizures, indicating that familial febrile seizures should also be considered a channelopathy.81
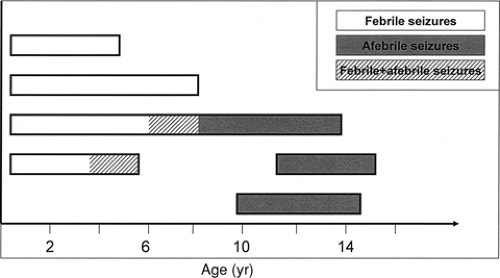 FIGURE 4. Clinical phenotypes in generalized epilepsy with febrile seizures plus (GEFS+). Range of phenotypes found in GEFS+ families is shown. Febrile seizure plus phenotypes include febrile seizures that persist beyond the age of 6 years and/or febrile seizures overlapped or followed by different types of (mostly generalized) afebrile seizures. (Adapted from Baulac S, Gourfinkel-An I, Nabbout R, et al. Fever, genes, and epilepsy. Lancet Neurol. 2004;3:421–430.) |
Generalized Epilepsy with Febrile Seizures Plus and Dravet Syndrome
Febrile seizures are the most common seizure type in families with the recently described syndrome of generalized epilepsy with febrile seizures plus (GEFS+) (see also Chapter 256), followed by febrile seizures plus (FS+; seizures with fever may persist beyond the age of 6 years and/or may be associated with variable afebrile seizures). Afebrile seizure types in affected GEFS+ individuals include generalized tonic–clonic, myoclonic, absence, and atonic seizures or, at least in some families, partial seizures105 (Fig. 4). Given the highly variable clinical phenotype, it is not surprising that the mode of inheritance underlying GEFS+ is still a matter of debate. Although in some families the trait is likely to be autosomal dominant, in others it is probably better described as oligogenic or as a major gene effect. A genetic concept involving more than one gene would also better fit the clinical variability observed within and between GEFS+ families. The first GEFS+ mutation (C121W) was identified in the SCN1B gene on chromosome 19q13.1.
SCN1B encodes a small accessory subunit of the voltage-gated sodium channel. The mutation C121W can be predicted to disrupt a disulphide bridge and to interfere with the ability of the β-subunit protein to modulate the function of the much larger, pore-forming α-subunits.140 Subsequent studies showed that most GEFS+ mutations are located in the gene coding for the α1-subunit of the voltage-gated sodium channel, SCN1A, on chromosome 2q24.41 SCN1A codes for a large ion channel protein composed of four domains, each containing six transmembrane segments (S1 to S6). Minor GEFS+ genes are SCN2A, coding for the γ2-subunit gene of the voltage-gated sodium channel, or the γ2-subunit gene of the GABAA receptor (GABRG2).8,56,66,82,123,139
SCN1B encodes a small accessory subunit of the voltage-gated sodium channel. The mutation C121W can be predicted to disrupt a disulphide bridge and to interfere with the ability of the β-subunit protein to modulate the function of the much larger, pore-forming α-subunits.140 Subsequent studies showed that most GEFS+ mutations are located in the gene coding for the α1-subunit of the voltage-gated sodium channel, SCN1A, on chromosome 2q24.41 SCN1A codes for a large ion channel protein composed of four domains, each containing six transmembrane segments (S1 to S6). Minor GEFS+ genes are SCN2A, coding for the γ2-subunit gene of the voltage-gated sodium channel, or the γ2-subunit gene of the GABAA receptor (GABRG2).8,56,66,82,123,139
Mutations in the SCN1A gene have also been identified in the majority of patients with the syndrome of severe myoclonic epilepsy of infancy (SMEI = Dravet syndrome; also Chapter 230) and in some cases of phenotypically overlapping syndromes like borderline SMEI (SMEB) or intractable childhood epilepsy with generalized tonic–clonic seizures (ICEGTC).31,45 The clinical phenotype of SMEI is characterized by an initially normal psychomotor development, onset of febrile seizures within the first year of life, and subsequent manifestation of various afebrile seizure types including absence, myoclonic, and partial seizures. During the second year of life psychomotor delay becomes evident.
Although caused by the same gene, SCN1A, differences in the mutation detection rates and the mutational spectrum present in GEFS+ and SMEI patients are obvious.87 SCN1A mutations are only found in 5% to 10% of GEFS+ patients but are present in 33% to 100% of SMEI patients (the variation is likely to be due to different mutation detection methods and/or patient inclusion criteria). All known GEFS+ mutations are missense mutations that cause amino acid exchanges in the encoded protein, while SMEI mutations are composed of truncating (nonsense or frame shift) mutations (47%), missense mutations (43%), splice site mutations (7%), and deletions (3%). Nearly all reported SMEI mutations (76 of 80) are de novo; only a few of them (4 of 80) were inherited from a parent with a less severe type of epilepsy. Mutations in SMEI tend to be either truncating and/or located in functionally critical parts of the gene. GEFS+ mutations are mostly found in less highly conserved parts of SCN1A such as the distal parts of S1 to S4 segments or in the short loops connecting those segments (Fig. 5). It is therefore likely that SMEI and GEFS+ mutations differ with respect to their predicted impact on ion channel function. SMEI mutations can be expected to cause a more severe disturbance of protein function, consistent with the more severe phenotype.
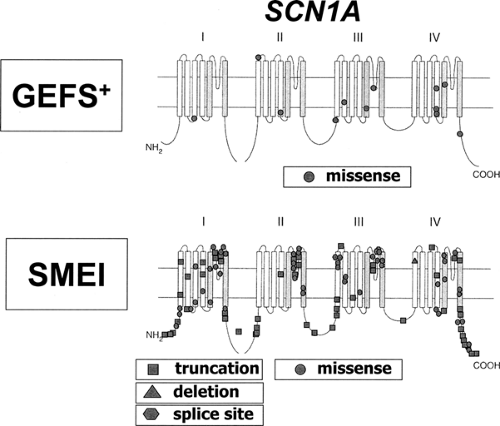 FIGURE 5. Schematic representation of SCN1A mutational spectrum in generalized epilepsy with febrile seizures plus (GEFS+) and severe myoclonic epilepsy of infancy (SMEI). All reported GEFS+ mutations are missense mutations. In SMEI different types of mutations are found, and most of these mutations are either truncating mutations or missense mutations affecting functionally important structures like the channel’s ion pore. (Adapted from Baulac S, Gourfinkel-An I, Nabbout R, et al. Fever, genes, and epilepsy. Lancet Neurol. 2004;3:421–430.) |
The exact functional effects of SMEI and GEFS+ mutations and their correlations to clinical phenotypes are still unclear. Expression studies of SCN1A channels with different SMEI and GEFS+ mutations in Xenopus oocytes or HEK293 cells showed a variety of biophysical aberrations including complete loss of function, altered gating properties, and even gain-of-function effects.77,78,101 These conflicting results can probably partially be explained by different experimental setups, since the biophysical properties of voltage-gated sodium channels strongly depend on the expression system used in the experimental setup. SCN1A channels expressed in HEK293 cells are known to differ from those expressed in Xenopus oocytes with respect to their kinetic properties and their sensitivity to β-accessory subunits. Furthermore, to be able to compare the different functional studies, the question needs to be addressed whether SMEI and/or GEFS+ mutations alter channel surface expression by affecting gene transcription, mRNA stability, protein folding, or trafficking.
Another syndrome of early childhood epileptic encepha- lopathy that is phenotypically and etiologically related to both SMEI and GEFS+ is ICEGTC. Both GEFS+ and ICEGTC are severe types of early childhood epilepsy that are characterized by fever sensitivity, intractable seizures, and developmental decline after seizure onset. Contrary to GEFS+, myoclonic or absence seizures usually do not occur in ICEGTC patients. SCN1A mutations were found in 7 of 10 ICEGTC patients. All mutations were missense mutations that were, similar to SMEI mutations, either located in highly conserved (S4 to S6, pore loop) or, as in GEFS+, less conserved parts of the gene (S1 to S4). Two of the seven published ICEGTC patients inherited their SCN1A mutations from a parent with GEFS+-like epilepsy, raising the possibility that additional genetic and/or environmental factors contribute to the more severe phenotype in ICEGTC.45
Novel Gene Families in Idiopathic Epilepsy
Most of the genes mentioned above either code for ligand-gated or voltage-gated ion channels (i.e., CHRNA4/B2, GABRG2, KCNQ2/3, SCN1A/2A) or for proteins that modulate ion channel function (i.e., accessory channel subunits). Recently, genes of so far unknown function belonging to newly identified gene families were found in some rare idiopathic epilepsies. The first of those was the LGI1 gene discussed below. These
findings demonstrate that “channelopathies” are not the only etiologic concept in idiopathic epilepsies, but that other mechanisms might exist.
findings demonstrate that “channelopathies” are not the only etiologic concept in idiopathic epilepsies, but that other mechanisms might exist.
Autosomal Dominant Lateral Temporal Lobe Epilepsy
The syndrome of autosomal dominant lateral temporal lobe epilepsy (ADLTE, also named autosomal dominant partial epilepsy with auditory features [ADPEAF]) is characterized by simple partial seizures with mainly acoustic and sometimes even visual hallucinations.144 The age of onset is usually in the second or third decade of life. In some families the seizures tend to start with a brief sensory aphasia without reduced consciousness.51 In 2002, positional cloning efforts let to the identification of the LGI1 gene (leucine-rich glioma inactivated gene 1) on chromosome 10q24 as the gene responsible for ADLTE/ADPEAF.44,51,65,85,100 The mutational spectrum includes both missense mutations and truncating mutations, and no mutation hotspot has emerged yet. There are no obvious clinical differences between patients with truncating mutations and those with amino acid exchanges in the LGI1 gene.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


