Fig. 17.1
T2-weighted transaxial (a) and coronal (b) imaging of the upper cervical spine shows marked atrophy, and there is extensive hyperintense gray matter signal. Note CSF flow artifact surrounding the spinal cord
17.3 Spinal Muscle Atrophy
Group of rare autosomal recessively inherited diseases linked to the same gene.
Age of onset and progression is determined by specific mutation.
Spinal MRI typically normal.
Spinal muscular atrophy (SMA) presents with progressive signs of lower motor neuron disease due to degeneration of the anterior horn of the spinal gray matter. Symptoms include symmetric muscular weakness with loss of deep tendon reflexes and muscular atrophy. Clinically and genetically, SMA is divided into four subtypes, all of which are autosomal recessive disorders caused by specific mutations of the SMN-1 gene. The type and location of mutation determine the age of onset, progression rate, and severity of symptoms. Disease onset can occur at any point in life, and life expectancy can be very limited with SMA type 1, where symptoms present within the first 6 months of life.
Imaging: Typically, MRI is normal in these patients, since the degeneration of the anterior horn of spinal gray matter is beyond the resolution of standard clinical scanning. With high-resolution imaging, however, degeneration can be detected (Fig. 17.2).
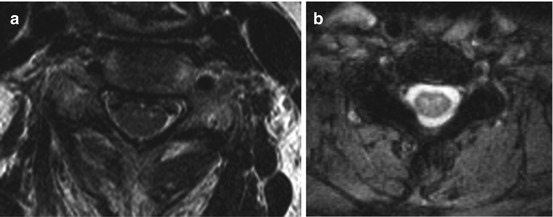
Fig. 17.2
High-resolution transaxial T2-weighted images show hyperintense signal in the anterior horn of the cervical (a) and thoracic (b) spinal gray matter
17.4 Poliomyelitis
Poliomyelitis, or polio, is caused by the poliovirus. Infection occurs via the fecal-oral route, and effective vaccines are available. Most patients with fresh infections will be asymptomatic or experience gastrointestinal symptoms only. In around 1–3 % of cases, however, the poliovirus enters the central nervous system. It targets spinal and bulbar motor neurons, leading to progressive weakness and muscular atrophy with loss of deep tendon reflexes. Clinically, motor symptoms are preceded by meningeal symptoms or even encephalitis. Diagnosis is made with stool cultures and serology.
The location of motor symptoms depends on the site of viral manifestation within the CNS, leading to spinal, bulbar, and bulbospinal forms of poliomyelitis. Bulbar involvement is particularly dangerous, leading to respiratory deficiency or palsy of pharyngeal muscles with subsequent dysphagia, or even fatal inflammation of the entire brain stem. Most symptomatic infections occur in children, and spinal polio can lead to deformities of the extremities due to insufficient musculature during the growth phase. The virus also targets neurons in the spinal ganglia and the deep cerebellar nuclei. Despite paresthesia during the initial phase, loss of sensation in the extremities is typically absent.
Note that some patients will experience progression of motor symptoms or progressive fatigue decades after the infection, a condition called post-polio syndrome that can be severely disabling.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


