CHAPTER 69 HEREDITARY SPASTIC PARAPLEGIAS*
The hereditary spastic paraplegias (HSPs), also known as familial spastic paraplegia and Strümpell-Lorrain disease (per Dorland’s),1 constitute a group of more than 30 inherited neurological disorders in which the predominant symptom is bilateral lower extremity spastic weakness. Previous reviews of HSP are available in articles by Fink and colleagues,2–4 in the Gene Reviews website (http://www.geneclinics.org/profiles/hsp/), in the University of Michigan’s Hereditary Spastic Paraplegia website (http://www.med.umich.edu/hsp), and in the website for the Spastic Paraplegia Foundation (http://www.sp-foundation.org).
EPIDEMIOLOGY
HSP affects individuals of all ethnic groups and ancestries without particular ethnic predilection. The prevalence of HSP has been estimated in Ireland (1.27 per 100,000),5 Italy (2.7 per 100,000),6 and Spain (9.6 per 100,000).5
GENETIC CLASSIFICATION: MODE OF INHERITANCE AND HSP LOCUS
There are autosomal dominant, autosomal recessive, and X-linked forms of HSP, each of which is genetically heterogeneous: Many different, separate gene mutations cause clinically similar, often indistinguishable syndromes of lower extremity spastic weakness). HSP genetic loci are designated SPG (spastic gait) and numbered 1 through 28 in order of their discovery (Table 69-1).

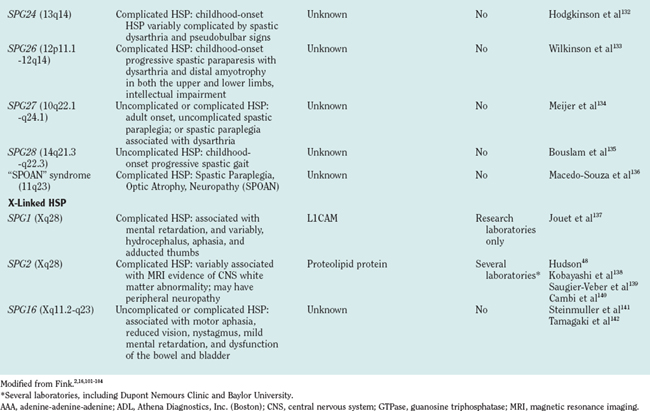
CLINICAL CLASSIFICATION: “UNCOMPLICATED” AND “COMPLICATED” HSP
Classifying HSP as uncomplicated or complicated is also important for prognosis. Families with “uncomplicated” HSP (e.g., HSP caused by SPG4/spastin gene mutation) are not at risk of having offspring with “complicated” HSP. The converse is not always true, however. Families with some forms of “complicated” HSP (e.g., SPG7 or SPG10 HSP caused by paraplegin or kinesin heavy chain [KIF5A] gene mutations, respectively)7,8 may have offspring affected with either “uncomplicated” or “complicated” HSP.
Controversies in HSP classification arise as the knowledge of the HSPs expands. For example, seizures, cognitive impairment, and ataxia9 have been reported in patients with the most common form of dominantly inherited HSP (SPG4, caused by SPG4/spastin gene mutation), generally considered prototypical of uncomplicated HSP.10–14 Further studies to determine the frequency of “extraspinal” features in otherwise uncomplicated HSP are needed.
NEUROPATHOLOGY: DISTAL AXON DEGENERATION INVOLVING LONGEST MOTOR AND SENSORY FIBERS IN THE CENTRAL NERVOUS SYSTEM
Postmortem studies of uncomplicated HSP reveal relatively selective axonal degeneration involving terminal portions of corticospinal tracts (maximal in the thoracolumbar region) and dorsal column fibers (maximal in cervicomedullary region).15–20 Spinocerebellar fibers are involved to a lesser extent. Myelin loss is considered secondary to primary axonal degeneration. Decreased numbers of cortical motor neurons and anterior horn cells have been reported.18,19 Peripheral nerves and dorsal root ganglia are normal in uncomplicated HSP.19
Axonal degeneration in uncomplicated HSP thus involves the distal ends of the longest motor (corticospinal tracts) and sensory (dorsal column) fibers in the central nervous system (CNS). In this regard, uncomplicated HSP may be considered a “CNS homologue” of Charcot-Marie-Tooth disease type 2, in which distal motor and sensory axon degeneration is limited to the peripheral nervous system. To extend this analogy, it is noteworthy that one type of HSP (SPG10) is caused by mutation in KIF5A,8 whereas mutations in another kinesin (KIF1B) cause Charcot-Marie-Tooth disease type 2A1.21
EMERGING CONCEPTS OF HSP PATHOGENESIS
The molecular mechanisms underlying axon degeneration for most types of HSP are poorly understood. Nonetheless, the discovery of many HSP genes is generating new concepts about the pathophysiology of HSPs.16 Thus far, 28 HSP loci and 11 HSP genes have been discovered (see Table 69-1). The functions of most of these HSP proteins have not been fully elaborated; however, as a group, HSP genes (and their respective proteins) appear highly diverse. This diversity of HSP genes (and their proteins) suggests that axonal degeneration in various genetic types of HSP may be caused by diverse, primary biochemical disturbances.22 A tentative biochemical classification of HSP is emerging. It is likely that these disparate biochemical disturbances converge into one or more common pathways.
HSP Caused by Axonal Transport Abnormality
There is increasing evidence that disturbance of axonal transport occurs in a variety of motor neuron disorders, including HSP.23,24 The clearest example of axonal transport involvement in HSP is autosomal dominant SPG10 HSP caused by KIF5A mutation. KIF5A is a molecular motor component involved in axonal transport of organelles and macromolecules. SPG4, the most common cause of dominantly inherited HSP, resulting from spastin gene mutation, may be another example of axonal transport or cytoskeletal involvement in HSP. There is accumulating evidence that spastin interacts with microtubules and may be involved in microtubule severing.25–34
HSP Caused by Golgi Abnormalities
Atlastin mutations cause approximately 25% of childhood-onset dominantly inherited HSP.35–37 Spartin mutations cause autosomal recessive HSP associated with distal muscle atrophy (Troyer’s syndrome).38 Although the functions of both atlastin and spartin have not been elucidated, it is known that both of these proteins are localized to the Golgi apparatus.39,40
HSP Caused by Mitochondrial Abnormality
Two HSP genes encode integral mitochondrial proteins: chaperonin 60/heat shock protein 60, mutations of which cause autosomal dominant uncomplicated SPG13 HSP,41 and paraplegin, mutations of which cause autosomal, complicated recessive SPG7 HSP.42–45 Some but not all patients with paraplegin mutation have evidence of mitochondrial abnormalities in skeletal muscle biopsy.7
HSP Caused by Primary Myelin Disturbance
It is important to note that axon degeneration in at least one form of HSP arises not from an intrinsic axon or neuron abnormality but rather from glial abnormality. X-linked SPG2 HSP is caused by proteolipid protein gene mutation. Proteolipid protein is an intrinsic myelin protein, and mutations in the gene cause both Pelizaeus-Merzbacher disease46 (an X-linked infantile-onset dysmyelination disorder) and X-linked HSP (a childhood-onset slowly progressive spastic gait disorder).47,48 Patients and gene-targeted mice lacking proteolipid protein develop length-dependent axon degeneration in the absence of demyelination.49
HSP Caused by Embryonic Development of Corticospinal Tracts
Mutations in the neuronal cell adhesion molecule L1 (L1CAM) gene cause a variety of X-linked neurological disorders, including X-linked hydrocephalus; the syndrome of mental retardation, aphasia, shuffling gait, and adducted thumbs; and complicated X-linked spastic paraplegia.50–52 L1CAM knockout mice53 exhibit weak hind limbs and reduced size of corticospinal tracts.
NEUROLOGICAL EXAMINATION: UPPER MOTOR NEURON SIGNS IN THE LEGS; IMPAIRED VIBRATION SENSATION IN THE TOES
Although all patients with HSP have lower extremity spasticity, the degree of weakness is variable. Some patients have lower extremity spasticity but normal muscle strength. Although Harding19,54 used the variable proportion of weakness and spasticity (along with age at symptom onset) to classify HSP as type I (greater spasticity than weakness) and type II (significant weakness), this classification is not widely used because estimates of the relative contributions of weakness versus spasticity are qualitative and because some genetic types of HSP may manifest as both type I and type II.
Vibration sensation in the toes is often mildly impaired in uncomplicated HSP. Distal lower extremity vibratory impairment may not manifest for several years, but when present, this is a helpful diagnostic sign. When not attributed to other disorders (such as peripheral neuropathy or cervical spondylosis), impaired vibration sensation in the toes helps to distinguish HSP’s motor (corticospinal tract) and sensory (dorsal column) patterns of involvement from primary lateral sclerosis (involvement of upper motor neurons with sparing of dorsal columns).55 Although vibratory sense may be mildly impaired in HSP, severe dorsal column disturbance is not typical of uncomplicated HSP and would prompt a search for other disorders (including Friedreich’s ataxia, subacute combined degeneration, and tertiary syphilis).
Spastic Gait
Patients with uncomplicated HSP generally exhibit bilaterally symmetrical gait disturbance,56 including short stride length (because of difficulty flexing the thighs and dorsiflexing the feet), circumduction, anterior-foot strike (tendency to walk on the balls of the feet or on the toes), scissoring, hyperlordosis, and sometimes hyperextension at the knee. The ability to walk on the heels is generally compromised. The abnormality of gait varies between individuals. Careful analysis of each affected individual’s gait is necessary to provide specific exercise recommendations, to determine whether the patient would benefit from spasticity-reducing medication, and to determine whether the patient would benefit from ankle-foot orthotic devices.
SYNDROME VARIABILITY
Within a given genetic type of HSP (such as SPG4 HSP caused by spastin gene mutation), there may be significant clinical variability. Part of this variability may result from the effects of different mutations.57 For example, whereas SPG4 HSP (caused by spastin mutation) is usually uncomplicated, ataxia in addition to spastic paraplegia has been reported in a family with SPG4 mutation GLN490Stop.9
Significant variability between affected patients who share the same HSP gene mutation reflects the influence of modifying genetic and possibly environmental factors. One source of modifying genes is polymorphisms in HSP genes themselves. Recently, Svenson and associates analyzed benign SPG4/spastin polymorphisms (S44L and P45Q) and showed that L44 and Q45 are each associated with a striking decrease in age at onset in the presence of the pathogenic mutations in SPG4/spastin’s adenine-adenine-adenine domain.58
Age at Symptom Onset
Age at symptom onset may be quite variable between different genetic types of HSP, between patients with one particular genetic type, and even within a family in which affected patients share the same HSP gene mutation. Although the average age at symptom onset is earlier for some types of HSP (SPG10, SPG3A, and SPG12)4 than for other types of HSP (SPG4, SPG13, SPG8, and SPG6), there is significant overlap in the range of ages at which symptoms begin. For SPG4 HSP, meta-analysis of 75 families did not reveal a correlation between spastin mutation class (missense, aberrant splicing, frameshift, premature truncation mutations) and age at symptom onset.59
Genetic Anticipation
Genetic anticipation has been reported in SPG4 HSP,60 including patients later shown to have point mutations (not trinucleotide repeat expansions) in the SPG4/spastin gene. The author and colleagues have observed apparent genetic anticipation in SPG3A HSP. For example, they identified the SPG3A mutation V253I in a 70-year-old patient who was asymptomatic and had normal neurological examination findings; his mutation-bearing son developed HSP in his 20s; and his mutation-bearing grandson developed HSP before age 7 (J. K. Fink, 2005 unpublished observation).
Relatively Nonprogressive versus Progressive Forms of HSP
When HSP begins after adolescence, symptoms usually progress steadily over many years. When symptoms begin in childhood, there may be very little progression even over 10 years.2
Syndromic Features
Although complicated forms of HSP have “syndromic” features (e.g., SPG9, SPG10, and SPG17 have motor neuropathy or distal wasting), such features may be present in only a minority of affected family members (e.g., some with SPG1061 and some with SPG745 had complicated HSP, whereas others had uncomplicated HSP).
Subclinical Cognitive Disturbance and Late-Onset Dementia
These features have been described in some but not all patients with the most common form of dominantly inherited HSP (caused by SPG4 mutations)10–14 and may be correlated with specific spastin mutations.62 Cognitive impairment is a feature of several forms (see Table 69-1) of complicated HSP, particularly SPG11, which appears to be the most common form of autosomal recessive HSP.
Severity
The extent to which HSP symptoms are disabling is quite variable both between different genetic types of HSP and within a given family.4
LABORATORY STUDIES, NEUROIMAGING, AND NEUROPHYSIOLOGICAL EVALUATION
Neuroimaging is important for ruling out alternative disorders, including multiple sclerosis, leukodystrophies, and structural abnormalities affecting the brain or spinal cord (see “Differential Diagnosis” section). Whereas brain magnetic resonance imaging in uncomplicated HSP is usually normal, those of complicated forms of HSP may reveal syndrome-specific abnormalities, such as thin corpus callosum in autosomal recessive SPG11 HSP (e.g., Casali et al63) and cerebral or cerebellar abnormalities in autosomal recessive SPG7 HSP. Magnetic resonance imaging of the thoracic spinal cord often demonstrates atrophy in uncomplicated HSP.9–11,64
Electromyographic nerve conduction studies usually yield normal results in uncomplicated HSP. Such studies are useful in ruling out other disorders (such as amyotrophic lateral sclerosis, Friedreich’s ataxia, Machado-Joseph disease). Such study results are usually normal in uncomplicated HSP.51,52,65 Exceptions have been reported, however, including peripheral neuropathy in some patients with SPG4 HSP caused by spastin frameshift mutation 906delT53 and axonal neuropathy associated with SPG3A HSP caused by atlastin mutation R495W.66 Subclinical sensory neuropathy in otherwise uncomplicated HSP has been described.67,68 Several types of complicated HSP are associated with peripheral neuropathy (see Table 69-1).
Somatosensory evoked potentials may demonstrate dorsal column impairment in uncomplicated HSP. Whereas somatosensory evoked potentials recorded from lower extremities often show delayed conduction, somatosensory evoked potentials recorded from the upper extremities are usually normal.52,69–72 This finding may help distinguish patients with autosomal recessive, uncomplicated HSP (and those with uncomplicated spastic paraplegia who do not have a family history of the disorder) from patients in an early stage of primary lateral sclerosis (in whom vibration sensation and dorsal column function are normal).55
Cortical evoked potentials provide a useful measurement of corticospinal tract conduction velocity. Studies of patients with uncomplicated HSP often reveal reduced conduction velocity and amplitude when cortical evoked potentials are recorded from the lower extremities.73–76 In contrast, cortical evoked potentials recorded from cervical spinal segments are usually normal or show only mildly reduced conduction velocity.75
< div class='tao-gold-member'>
Related posts:
 AUTISM AND ATTENTION DEFICIT/HYPERACTIVITY DISORDER
AUTISM AND ATTENTION DEFICIT/HYPERACTIVITY DISORDER
 PRINCIPLES OF NEUROPSYCHOMETRIC ASSESSMENT
PRINCIPLES OF NEUROPSYCHOMETRIC ASSESSMENT
 TRIGEMINAL AUTONOMIC CEPHALALGIAS: CLUSTER HEADACHE AND RELATED CONDITIONS
TRIGEMINAL AUTONOMIC CEPHALALGIAS: CLUSTER HEADACHE AND RELATED CONDITIONS
 ANATOMY AND PHYSIOLOGY OF CEREBRAL AND SPINAL CORD CIRCULATION
ANATOMY AND PHYSIOLOGY OF CEREBRAL AND SPINAL CORD CIRCULATION
 TUMORS OF THE SPINAL CORD
TUMORS OF THE SPINAL CORD
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


