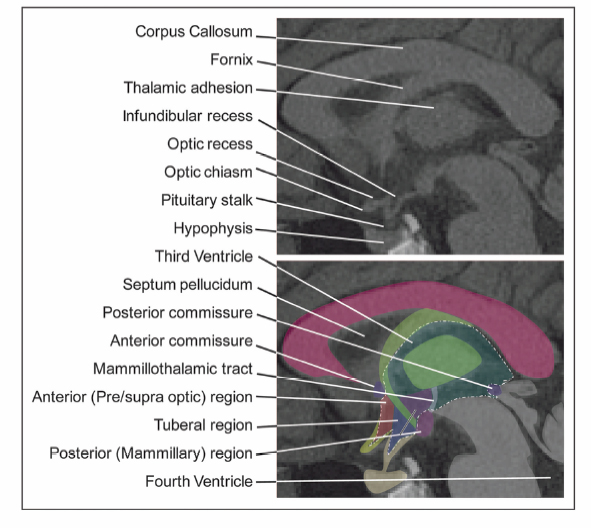
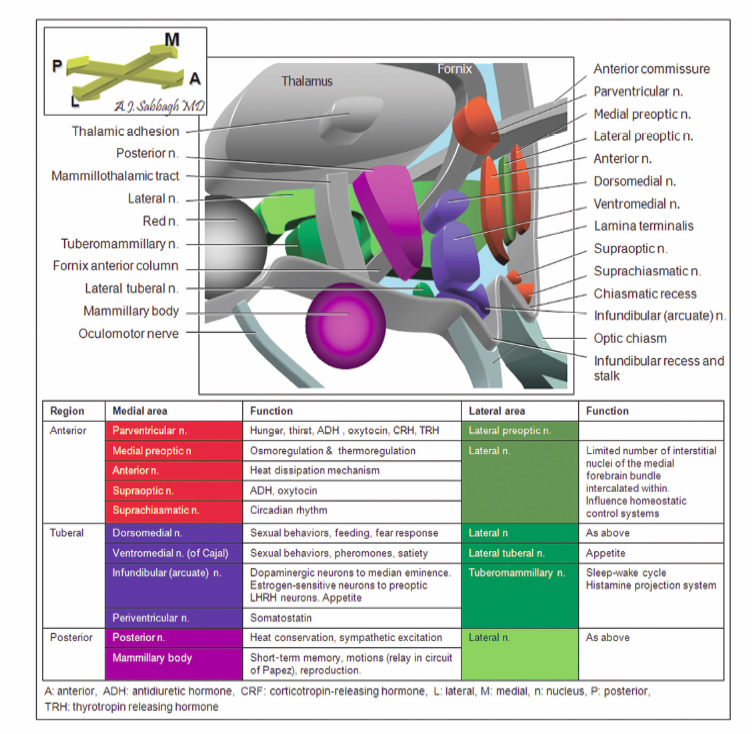
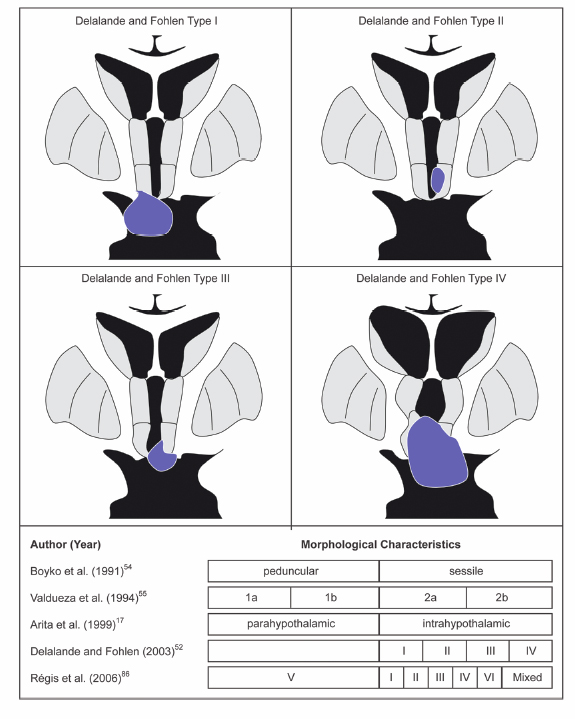
9 Hypothalamic Hamartomas The term hamartoma is derived from the Greek word hamartion meaning bodily defect. A hamartoma is a focal benign growth in an organ composed of tissue elements normally found at that site but that are growing in a disorganized mass. Hamartomas result from an abnormal formation of normal tissue, although the underlying reasons for the abnormality are not fully understood. Although hamartomas resemble neoplasms, they do not show any particular tendency for neoplastic evolution. Instead, they grow along with, and at the same rate as, the organ from whose tissue they are made, and, unlike cancerous tumors, only rarely invade or compress surrounding structures significantly. Hypothalamic hamartomas (HHs) are rare, heterotopic masses consisting of an abnormal mixture of neuronal and glial cells. They arise from the floor of the third ventricle, tuber cinerium, or mammillary bodies. These developmental malformations can be diagnosed incidentally during routine autopsy. They are often associated with a range of neurological and endocrine disturbances. In its most disabling form, a progressive epileptic encephalopathy appears in early childhood. In addition to gelastic seizures, which represent the hallmark of HH-related epilepsy, the syndrome is also commonly characterized by other types of medically refractory seizures. As well, patients with HH typically display severe cognitive impairment, pervasive developmental disorders, behavioral disturbances, and psychiatric disorders. A distinct subtype of patients present with central precocious puberty. The hypothalamus is the single most integrative structure in the cerebrum. It lies at the base of the brain, around the third ventricle, extending from a plane immediately anterior to the optic chiasm to one immediately posterior to the mammillary bodies. Its weight in the adult human is less than 2.5 g.1 In midsagittal section, the human hypothalamus is bound anteriorly by the lamina terminalis, posteriorly by a plane drawn between the posterior commissure and the caudal limit of the mammillary body, and superiorly by the hypothalamic sulcus. Ventrally, the hypothalamus encompasses the floor of the third ventricle, the inferior surface of which is termed the tuber cinerium (gray swelling). Laterally, the border is somewhat ill-defined, and the hypothalamus is roughly bounded by the internal capsule, cerebral peduncle, optic tract, and subthalamus. The hypothalamus can be divided into two longitudinal zones: a densely cellular medial zone and a relatively paucicellular lateral zone roughly separated by a sagittal plane passing through the anterior columns of the fornix. In humans, the majority of named hypothalamic nuclei are in the medial zone. In addition, the hypothalamus is commonly subdivided into regions along its anteroposterior axis (Fig. 9.1). The preoptic region extends rostral to the optic chiasm and dorsally to the anterior commissure. The supraoptic region resides above the optic chiasm. The tuberal region lies above and includes the tuber cinerium. Finally, the mammillary region includes the mammillary bodies and the posterior hypothalamic nuclei. Through its numerous reciprocal afferent and efferent connections, the hypothalamus coordinates autonomic, somatic, endocrine, and behavioral activities of the body. It receives input and in turn projects to a wide distribution of neurons arising in the forebrain, brainstem, and spinal cord. Figure 9.2 summarizes the major hypothalamic nuclei, their location, and function. Interestingly, the hypothalamus is also implicated in laughter, the behavioral manifestation of mirth.2,3 The embryological origin of HHs remains unknown. The true incidence of HH is indefinite, but the estimated prevalence is 0.5 per 100,000.4 These lesions are usually sporadic, and their exact etiology has not been determined. However, rather than presenting as isolated masses, HHs can be part of a multiple malformation syndrome. In fact, there are several congenital anomaly syndromes in which HHs have been described. Syndromic HHs generally have milder symptoms than do nonsyndromic sporadic HHs, but likely arise from similar pathogenetic mechanisms. The heritable nature of these disorders means that the genes that are mutated in affected patients can be detected. The identification of genes that, in the mutant state, cause HHs, suggests that the normal function of these genes are important for the usual development and function of the hypothalamus. Fig. 9.1 Midsagittal diagram of the hypothalamus depicting the surrounding neural structures at the level of the anterior columns of the fornix. Pallister-Hall syndrome (PHS), a developmental disorder first described in 1980 in a cohort of six children, is the most extensively studied disorder in terms of the molecular pathology.5 The syndrome is typically characterized by the presence of an HH in association with multisystem malformations. The spectrum of features also includes central postaxial polydactyly, pituitary hypoplasia or dysfunction, dysplastic nails, bifid epiglottis, and imperforate anus. In some cases, cardiac anomalies, renal defects, and mild mental retardation are seen. Given this characteristic phenotype, PHS is often diagnosed at birth. In familial cases, PHS has been noted to be inherited in an autosomal dominant pattern with variable expressivity. It has been linked with a mutation to a zinc finger transcription factor gene, Gli3, which resides on chromosome 7p13 in some inherited probands.6 Gli proteins act downstream of the sonic hedgehog (SHH) signaling pathway to regulate target gene expression. SHH plays a critical role in regulating dorsoventral patterning of the developing central nervous system.7 In the presence of SHH, the Gli3 transcription factor is released intact and migrates to the nucleus to activate downstream genes. In the absence of SHH, Gli3 appears to be processed by a protease into a shorter form that represses downstream genes. In patients with PHS, frameshiftmutations in the Gli3 gene cause the production of a truncated version of the protein, which is functionally identical to the processed, repressor form of the protein.8 Thus, the pathogenetic mutations in patients with PHS remove the ability of SHH to switch Gli3 between the repressor and activator state. There appears to be a correlation between the clinical features of HHs and their physical appearance in relation to the normal hypothalamic and surrounding structures. In addition to the classic presentation of gelastic seizures, patients with HHs also commonly develop multiple other seizure types that evolve with age. Other associated symptoms can include precocious puberty, behavioral disturbances, and progressive cognitive decline. The phenomenon of “pressure to laugh” or gelastic epilepsy was first described by Trousseau in 1873.9 Gelastic seizures, the hallmark of epilepsy related to HHs, typically begin in early childhood, often in the neonatal period, and are invariably followed by development of medically refractory catastrophic seizures. Gelastic fits are characterized by recurrent brief seizures with initial emotionless laughter or grimacing. In addition to ictal laughter, other seizure types commonly occur, which are often more disabling than the gelastic spells.10,11 Nevertheless, the vast majority of electrophysiological studies have focused on gelastic seizures. Arzimanoglou and colleagues12 reviewed 16 children with HH and intractable seizures. They found a wide clinical spectrum and variable severity of the epilepsy syndrome. This was in keeping with the findings of Frattali et al13 and Leal et al14 who also reported the presence of multiple seizure types, such as generalized tonic-clonic seizures, partial complex seizures, drop attacks, and atypical absences, in the majority of patients. Tassinari et al15 published the most extensive review of patients with gelastic seizures. The first part of their chapter reported on 60 patients with gelastic seizures without the presence of an HH, suggesting that ictal laughter is not pathognomonic of HH. The second part of the review detailed the profile of another 60 patients with gelastic seizures related to HH. In 51 patients of the latter group, gelastic seizures were the first seizure type to be observed. However, 75% of the patients had other types of intractable seizures, including complex partial seizures with or without secondary generalization in 35.5%, “falling” seizures in 33.3%, tonic seizures in 17.7%, and tonic-clonic seizures in 15.1%. Mullatti16 and Castro et al10 observed similar findings in adults with HHs. Castro et al observed two major seizure types other than gelastic seizures in six adults with HH: complex partial seizures and tonic seizures with or without secondary generalization.10 Mullatti studied 14 adult patients with HH and epilepsy.16 Only three patients developed seizures in adult life. She concluded that later onset of epilepsy seems to be associated with a milder epilepsy syndrome with less prominence of gelastic seizures. In addition, cognitive and behavioral difficulties were less common in adults compared with children with HH. An almost constant feature in patients with HH and pharmacoresistant gelastic seizures is development of a severe epileptic encephalopathy and catastrophic epilepsy of childhood. Often this epileptic syndrome is accompanied by profound behavioral problems, with reports of hyperactivity, rage and aggression being common.17–19 In addition to delinquency and aggressive outbursts, a slowly progressive deterioration in cognitive functions has also been frequently observed in patients with gelastic seizures and HH.11,12,20 Worsening of the behavioral and cognitive deficits usually parallels the aggravation of epilepsy. In Tassinari’s review, mental impairment was reported in 49 of 60 patients (81.6%) with gelastic seizures and HH.15 In addition, behavioral disturbances were present in 34% of the cases. Arzinmanoglou et al reported progressive development of cognitive and behavioral deterioration in all 16 children.12 Frattali et al reported mild to severe cognitive deficits in all eight children with HH and refractory epilepsy.13 Similarly, Mullatti et al reported mild to severe learning difficulties in 14 of 16 patients with childhoodonset seizures.11 Quiske et al published the first detailed report on cognitive performance in juvenile and adult patients with gelastic seizures and HH.19 They performed a comprehensive neuropsychological assessment and concluded that more than half of the 13 patients displayed deficits in a broad range of cognitive functions. Most prominent in were impairments in working memory, visual and verbal learning and memory (77% and 62%, respectively), and limitations in global intellectual performance (54%). A considerable number of patients showed below-average functions in cognitive flexibility and speed, concentration, and visuoconstructive abilities. Frattali et al found a significant relation between partial seizure frequency and broad cognitive ability score.13 Fig. 9.2 Summary of the major hypothalamic nuclei and their function. There is also a high prevalence of psychiatric comorbidity in patients with HH.21 Weissenberger et al evaluated 12 children between 3 and 14 years of age, along with parents and age-matched siblings.18 They found that children with HH and gelastic seizures had a significantly higher psychiatric morbidity, such as oppositional defiant disorder (83.3%), attention deficit-hyperactivity disorder (75%), conduct disorder (33.3%), and affective disorders (16.7%). Ali et al looked at 10 adult patients with HH and a history of intractable seizures.22 They also documented a high prevalence of comorbid mood and anxiety disorders with major depressive disorder and social anxiety disorder being the most common. The association between HH and central precocious puberty (CPP) is well established. Pedunculated HH below the third ventricle is rarely associated with seizures but may present with CPP defined as the onset of puberty before age 8 in girls and 9 in boys. CPP tends to occur significantly earlier than idiopathic CPP.23 The first manifestation of CPP in patients with HH occurs before age 2 in 82% of cases.23 Several hypotheses have been formulated to explain the potential mechanisms by which HHs cause precocious puberty.24 One hypothesis suggests that HHs accelerate sexual development by producing bioactive substances that mimic, in an accelerated time-course, the cascade of events underlying the normal initiation of puberty. Alternatively, it has been proposed that because HHs contain key transcriptional and signaling networks required to initiate and sustain a pubertal mode of gonadotropin-releasing hormone (GnRH) release, they are able to trigger the pubertal process at an earlier age. Finally, it is also possible that the developmental abnormalities leading to the formation of HHs result from sporadic defects affecting the same genes and hence the same morphogenic pathways involved in the embryonic development of the ventral hypothalamus and the floor of the third ventricle. The exact mechanisms involved in HH causing CPP remains to be fully elucidated. Patients with HHs typically have unremarkable skull x-rays. However, these slowly growing expansile masses can result in local bony erosion at the tip of the dorsum sella. Lin et al (1978) described suprasellar calcification in a patient harboring a histologically proven hamartoma of the tuber cinerium.25 Diebler and Ponsot (1983) reported a case in which detachment and anterior displacement of the tip of the dorsum sella was seen mimicking a calcified suprasellar craniopharyngioma.26 For historical interest, pneumoencephalography was a sensitive study to define midline lesions and often disclosed a small filling defect in the tip of the infundibular recess (Takeuchi et al, 1979).27 Although an angiographic study has little or no diagnostic value, it may demonstrate slight posterior displacement of the distal basilar artery and anterior pontomesencephalic vein, upward displacement of the A1 segment of the anterior cerebral arteries, and possibly lateral displacement of the posterior communicating arteries. Tumor blush or associated abnormal vessels are typically not encountered. The computed tomography (CT) findings are dependent on the size of the hamartoma. Plain CT shows a mass in the interpeduncular and suprasellar cistern, which has the same density as normal brain and does not enhance following contrast administration. Obliteration of the suprasellar cistern and anterior third ventricle is commonly observed.28 Metrizamide CT cisternography showed a filling defect in the suprasellar cistern and greatly improved the diagnostic yield for smaller occult lesions of the hypothalamic-hypophyseal axis by clearly defining the size of the mass as well as its relationship to surrounding neurovascular structures.29 The incidence of HHs has increased since the introduction of high-resolution magnetic resonance (MR) imaging. The detailed imaging of the lesion and its relationship to adjacent structures is of paramount importance in the planning of the surgical approach and during the procedure. Highresolution MRI scanning has therefore largely supplanted all other diagnostic studies and remains the neuroimaging modality of choice for evaluation of patients with lesions of the tuber cinerium. Freeman et al systematically studied the MRI findings of 72 patients with HH and refractory epilepsy.30 The vast majority of HHs (93%) showed increased T2-weighted signal intensity seen relative to cortical and deep gray matter. Unlike previous studies, which reported signals isointense with gray matter on T1-weighted images,31–33 the authors found that 53 cases (74%) were hypointense compared with the normal gray matter. The authors explain that this observation may be due to the greater signal intensity contrast among gray matter, white matter, and abnormal tissue obtained with inversion-prepared gradientecho sequences, compared with conventional spinecho sequences. The HH did not show contrast enhancement following administration of gadolinium, thereby demonstrating the integrity the blood-brain barrier. Magnetic resonance spectroscopy imaging is a highly sensitive method of detective neuronal dysfunction in patients with temporal lobe epilepsy. However, very few studies have reported the spectroscopic properties of HHs. Seven studies with a total of 42 patients underwent magnetic resonance spectroscopy with evaluation of the N-acetylaspartate (NAA), creatine (Cr), choline (Cho), and myoinositol (ml) content within the hamartoma. The group in Montreal was the first to study hypothalamic hamartomas using proton MR spectroscopic imaging.34 They quantified the amount of neuronal damage in the temporal lobes and hamartomas of five patients with HHs and gelastic seizures. The relative intensity of NAA/Cr was determined for both temporal lobes as well as for the hamartoma. These values were compared with signals from the temporal lobes and hypothalami of normal control subjects. The NAA/Cr ratio was not significantly different from normal control subjects for either temporal lobe, nor was there a significant asymmetry between the two temporal lobes for any of the patients. In contrast, NAA resonance signals were present in the hamartomas, and the ratio of NAA to Cr was significantly decreased in the hamartomas compared with the hypothalami of normal control subjects. This study provided further evidence that gelastic seizures do not originate in the temporal lobes of patients with HHs. Similarly, Pascual-Castroviejo et al used MR spectroscopy in their patient with an HH to localize and measure in the temporal lobes and in the lesion.35 The relative intensity of NAA to Cr and NAA to Ch was not significantly different from normal control subjects for either temporal lobes. Within the hamartoma, the ratio NAA/Ch was decreased, however, contrary to the earlier report of Tasch et al,34 the ratio NAA/ Cr was highly increased in the hamartoma in their patient with intractable gelastic seizures.35 The altered chemical shiftimaging with MR spectrometry in three other patients suggested a biochemical abnormality in the tissue of the HH. Wakai et al36 found a significant reduction of the NAA/serum creatinine ratio. Wakai et al further demonstrated decreased NAA and increased choline and creatine in the hypothalamic hamartoma. Martin et al found on short-TE proton MR spectra, the NAA concentration in the hamartoma to be lower than in the thalamus but similar to that in the amygdala.37 In addition, the myoinositol concentration was elevated in the hamartoma compared with that in the amygdala and thalamus. Recently, two groups reported a larger series of patients with HHs evaluated with MR spectroscopy. In 2004, Freeman and coworkers performed proton MR spectroscopy of the HH in 19 patients and compared it with the metabolite profile of the thalamus in 10 normal children and the frontal lobe in 10 normal adults.30 They also found reduced NAA and increased myoinositol content. Finally, Amstutz and colleagues studied data from single voxel spectroscopic sequences in 14 patients.38 They demonstrated a statistically significant decrease in NAA/Cr and an increase in mI/Cr ratios in tumor tissue when compared with values in normal gray matter of the amygdala. In addition, Cho/Cr ratios were also increased when compared with those in normal gray matter controls. Overall, these results from MR spectroscopy suggest that HHs manifest reduced neuronal density and relative gliosis compared with normal gray matter. The association between the typical severe cases of HHs and epilepsy was obscured by the observation of slow spike and wave electrograph (EEG) patterns with or without multifocal (typically frontal or temporal) epileptiform abnormalities.31 In addition, some seizures associated with HHs can resemble complex partial seizures of temporal or frontal origin both clinically and with ictal electroencephalography (EEG). Even with ictal recordings with intracranial electrodes (not including the HH itself), the frontal or temporal cortex can appear to be the site of origin of seizures.39 However, neocortical resection is uniformly ineffective in controlling the seizures.39 An ictal EEG correlate is frequently lacking during gelastic seizures.40 This is consistent with ictal onset in a deep-seated structure. Ictal activity may not be captured by scalp electrodes, but it can be seen in the HH when depth electrode recording is used. Munari’s group in Grenoble performed the first depth electrode recording directly from the HH in a patient. During the same time period, our group in Montreal implanted electrodes within the HH in 2 patients. These landmark studies provided direct evidence that the ictal events, specifically, the gelastic seizures, are linked to discharge that is confined to the hamartoma itself.41,42 Over the last decade, ictal EEG recordings derived from a relatively small number of patients who underwent electrode placement within the HH have further confirmed that the ictal events associated with gelastic seizures indeed arise directly from the HH itself.43–45 Furthermore, electrical stimulation of the depth electrode contacts inserted into the HH lesion provoked the habitual seizures in some of these patients.41,43,44 Likewise, Choi and colleagues stereotactically implanted depth electrodes within the HH and confirmed the HH-induced epileptic discharges.45 Interestingly, they observed immediate disappearance of these epileptic discharges on intraoperative or postoperative EEG monitoring following endoscopic disconnection. Clear evidence for the intrinsic epileptogenesis of the hamartoma was further reinforced by metabolic studies. Palmini et al reported one patient who underwent an interictal [18F]-fluoro-deoxyglucose (FDG) PET scan that showed focal cortical hypometabolism colocalizing with the most prominent scalp EEG abnormalities.42 Ryvlin et al investigated brain glucose metabolism in patients with epileptogenic HHs, in an attempt to identifysigns of focal cortical and subcortical dysfunction that might correlate with other clinical data.46 They studied five patients with epileptogenic HHs using FDG-PET. The anatomical distribution of interictal FDG-PET abnormalities was compared with that of interictal and ictal electroclinical findings. All five patients demonstrated focal hypometabolism grossly concordant with the cortical regions suspected to participate in the ictal discharges. Epileptogenic HHs are usually associated with focal cortical hypometabolism in regions that might participate in the overall hamartomadriven epileptic network. Whether these cortical abnormalities only reflect the propagation of ictal discharges or a potentially independent seizure onset zone remains unknown. Further insight into the intrinsic epileptogenicity of HHs was gained by the study of Palmini and colleagues.47 They reported the first ictal FDG-PET evidence of the hypothalamic origin of gelastic seizures in a patient with an HH. Ictal FDG-PET was acquired during an episode of status gelasticus with preserved consciousness, in a patient previously operated on for complex partial seizures due to a temporal lobe epileptogenic cyst. Ictal hypermetabolism was localized to the region of the hamartoma during the status gelasticus. This elegant ictal FDG-PET study independently confirmed that gelastic seizures in patients with HHs do indeed originate in the diencephalic mass. Further evidence identifying the HH as the focus of seizure generation has been provided by measurements of regional cerebral blood flow during gelastic seizures in patients with HHs. Several studies performed ictal SPECT scans after injection of the tracer [99m]technetium hexamethylpropyleneamine oxime (Tc-HMPAO). These reports consistently demonstrated dramatic ictal hyperperfusion within the hamartoma with normalization during the interictal phase lending further support to the notion that certain types of HHs are inherently epileptogenic in nature.43,48–53 Whether the intrinsic epileptogenicity of HHs is responsible for the entire clinical spectrum of epileptic, neuropsychological, and behavioral disorders associated with HH, remains an open issue, in as much as morphologically similar HH can be associated with dramatically different seizure types and cognitive outcomes. A distinction must be made between the two different anatomical subtypes of HHs. The first subtype, referred to as the intrahypothalamic or sessile HH, consists of lesions whose base of attachment within the third ventricle is either partial or complete. These lesions vary significantly in size but typically extend into the third ventricle itself and distort local anatomical structures, including the fornix and mammillary body.30 Sessile HHs are highly associated with neurological symptoms, including gelastic seizures.54,55,17,40,56 These lesions typically do not present with precocious puberty, although up to 40% of these patients will develop CPP at some point during their course. The second subtype is referred to as the parahypothalamic, or pedunculated HH. These lesions are attached only to the floor of the third ventricle or suspended from it by a peduncle. In contrast to sessile lesions, pedunculated HHs typically do not present with a catastrophic childhood epilepsy syndrome or neurodevelopmental disorder. Rather, they are commonly associated with CPP as the presenting abnormality. As discussed previously, the interference of HHs with inhibitory pathways onto GnRH neurons has been hypothesized as a possible mechanism for CPP. It is therefore conceivable that HHs are more likely to be associated with CPP if they are located close to the GnRH nerve terminals in the median eminence, as in a parahypothalamic position. Resection of pedunculated lesions causing precocious puberty reverses the hormonal abnormalities.57,58 However, medical therapy using leuprolide acetate (a long-acting GnRH analog) alone is now the first-line treatment for most of these patients. The majority of HHs can be easily classified as either sessile or pedunculated. However, some intermediary forms have been described; for example a large sessile HH that has a broad base of attachment to the underside of the hypothalamus and that extends into the interpeduncular cistern. According to Valdueza et al,55 epilepsy in HHs is observed only in medium and large sessile HH broadly attached to the tuber cinerium or mammillary body. The hypothesis that size, shape, and location of HHs affect the likelihood of clinical presentation is compelling. Jung et al40 reported that hamartomas inducing gelastic seizures tend to be larger (≥10 mm in diameter) than those with isolated CPP. Sturm et al59 observed that some patients with small HHs showed pressure to laugh, often without actual laughter, and suggested a dose–response relationship between the size of the HH and the severity of the epilepsy. However, because a significant proportion of patients with a HH of ≥10 mm in diameter presented with isolated sexual precocity, Jung and colleagues40 emphasized that size cannot be the only characteristic responsible for the induction of these clinical symptoms. Figure 9.3 outlines the different anatomotopographical classifications of HHs based on MRI findings reported by various authors. The major breakthrough in our understanding of the pathophysiology of HH was the discovery that HHs have intrinsic epileptogenicity. It is believed that through their mamillothalamic tract connections, these lesions predispose to generalized seizures and generalized interictal spike and wave discharges. Until recently, the histopathology of HH was poorly understood. Initial analysis showed that the hamartomas were composed of disorganized networks of neurons and glia similar to the structural lesions associated with epilepsy such as cortical dysplasia. Immunohistochemistry showed that these neurons stained intensely for NeuN, a marker for mature neurons, and were surrounded by a fine fibrillary matrix positive for synaptophysin immunostaining. Immunocytochemistry with glial fibrillary acidic protein (GFAP) showed scanty astrocytes and oligodendrocytes without neoplastic differentiation.55 In a recent article, Coons et al60 provided detailed histopathological analysis of 57 cases of HH. They noted that a consistent feature of all HH specimens examined was the presence of neuronal clusters or nodules that varied in size. The neuronal nodules were composed predominantly of small bipolar neurons interspersed with and surrounded by large pyramidal neurons in an astrocyte-rich neuropil.
Topographic and Functional Anatomy of the Hypothalamus
Etiology and Molecular Biology of Hypothalamic Hamartomas
Clinical Characteristics
Epileptic Syndromes
Behavioral and Cognitive Disturbances
Psychiatric Manifestations
Precocious Puberty and Other Endocrine Dysfunction
Neuroimaging Findings
Plain X-rays
Pneumoencephalography
Cerebral Angiography
CT Scan
MR Imaging
MR Spectroscopy
Neurophysiology and Intrinsic Epileptogenesis
Depth Electrode Recording
Positron Emission Tomography (PET) Scan
Single Photon Emission Computerized Tomography (SPECT) Imaging
Morphological Characteristics and Topological Classification
Neuropathological, Neurobiological, and Neurophysiological Findings
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


