9 Hypoxic Ischemic Encephalopathy
Adam Webb and Owen Samuels
More than 160,000 out-of-hospital cardiac arrests occur annually. Nearly 25% of patients receive cardiopulmonary resuscitation (CPR) either from bystanders or emergency services (EMS) personnel and many regain spontaneous circulation.1 However, despite modern improvements in CPR and critical care, less than 10 to 20% will survive to hospital discharge.2 Of those that do survive, many will have profound neurologic injury and disability.
Hypoxic-ischemic encephalopathy is the term used to describe brain injury after cardiac arrest, severe hypoxia, or prolonged hypotension. It shares some pathophysiologic mechanisms with other causes of global cerebral injury, such as respiratory arrest, severe hypoglycemia, and carbon monoxide poisoning.
After cardiac arrest, brain oxygen supply is depleted, setting off a cascade of injury. Within minutes, adenosine triphosphate (ATP) supplies are depleted, and energy-dependent membrane ion transport begins to fail. The excitotoxic neurotransmitter glutamate is released, and intracellular calcium accumulates activating proteases and phospholipases. A second period of injury begins when spontaneous circulation is restored. Several mechanisms contribute to reperfusion injury, including the formation of damaging oxygen free radicals and impaired autoregulation of cerebral blood flow. Initially after reperfusion there is rebound hyperemia followed by hypoperfusion, which is exacerbated by impaired cerebral microcirculation.
Specific areas of the brain are selectively vulnerable to global ischemia, including CA1 neurons of the hippocampus, cerebellar Purkinje cells, and pyramidal neurons in the cerebral cortex (layers 3, 5, and 6). Watershed regions between major cerebral vascular territories are also at risk for injury.
History and Examination
History
Review the EMS record or interview any witnesses to determine how much time elapsed between the time the patient collapsed and initiation of CPR. Did the patient receive any medications during resuscitation or intubation that might influence your exam (atropine, sedatives, neuromuscular blocking agents, etc.)?
- Vital signs: Assess blood pressure, temperature, heart rate and rhythm, spontaneous respirations, breathing pattern, and oxygen saturation.
- Evidence of trauma: Remember that if the patient is found down, always assume trauma is a possibility, and immobilize the cervical spine.
Neurologic Examination
- A full neurologic examination, including assessment of mental status, cranial nerves, motor skills, and reflexes, as well as a sensory and cerebellar exam, should be performed on all patients.
- Level of consciousness: Verbal responses, eye opening, response to pain
- Cranial nerves: Pupillary responses, oculomotor responses, cold water caloric responses, corneal reflex, grimace to nasal stimulus or supraorbital pressure, gag and cough reflexes
- Motor skills: Tone, motor responses to sensory stimulus, spontaneous or purposeful movements, motor posturing
Differential Diagnosis
- Cardiac arrest. Review the history of the arrest and rhythm strips. It is important to determine the type of arrest (cardiac origin: ventricular fibrillation or ventricular tachycardia; metabolic or pulmonary origin: pulseless electrical activity, asystole, bradycardia).
- Respiratory arrest
- Profound hypoglycemia. Assess for history of liver or renal failure, iatrogenic hypoglycemia, medication overdose.
- Drug overdose or toxic ingestion. Review medication access, perform a toxicology screen.
- Trauma (especially if history is not well established)
- Nonconvulsive status epilepticus. When in doubt, check an electroencephalogram (EEG).
- Stroke. A stroke can induce cardiac arrhythmias, and cardiac arrest can cause a focal stroke, especially in a patient with underlying cerebrovascular disease. Keep a high degree of suspicion in patients with focal findings on exam. Always test vertical eye movements to command to rule out a locked-in syndrome. Bilateral anterior cerebral artery (ACA) infarcts can produce an akinetic mute state that may mimic a persistent vegetative state.
- Other metabolic derangements that may cause both cardiac and cerebral dysfunction (i.e., acute renal failure)
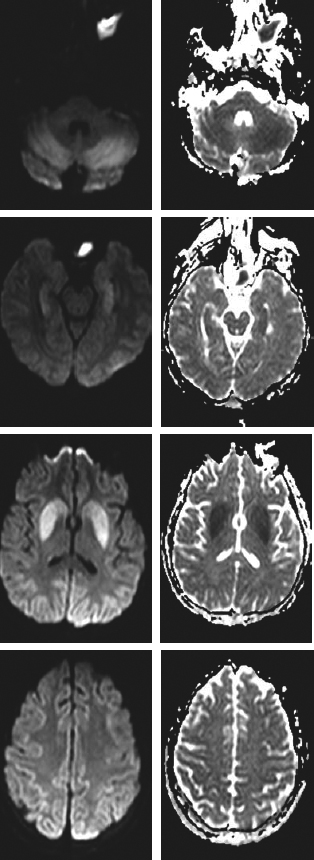
- Carbon monoxide poisoning. Look for pallor (more common) or cherry red skin (late sign), bright red retinal veins (early sensitive sign), and a history of exposure. Typically, the PaO2 is normal, but O2 saturation is low. Use co-oximetry to check for elevated carboxyhemoglobin levels (levels normalize if enough time from exposure has lapsed, and chronic smokers may have levels of CO as high as 10%). Magnetic resonance imaging (MRI) may reveal bilateral globus pallidus necrosis or cerebral edema. If neurologic or cardiac dysfunction is present or CO is >40%, hyperbaric O2 is indicated, otherwise 100% O2 is the therapy of choice.
Diagnostic Evaluation
- Laboratory studies. Serum glucose, chemistry profile to look for reversible causes of encephalopathy, drug screen, consider serum NSE (neuron specific enolase) and S100, cerebrospinal fluid creatine kinase BB activity (CSF CKBB; if available)
- Imaging studies
- Noncontrast head computed tomography (CT):
- Used to exclude primary brain injury as a cause of both coma and cardiac arrest
- Often normal following cardiac arrest
- May see obscuration of gray-white junction, watershed infarcts, or abnormal appearance of deep gray matter nuclei
- MRI:
- Neurophysiologic studies
- EEG:
- Somatosensory evoked potentials (SSEPs):
- Provide a bedside means of assessing brainstem and cortical function
- Less affected by medications and metabolic derangements than EEG
- Bilateral absence of N20 response with median nerve stimulation is strongly predictive of poor outcome; however, almost 50% of patients with present N20 will also have a poor outcome.3
- Timing is important: N20s may initially be absent and then return. Optimal timing is 1 to 3 days from arrest.
- SSEPs can be difficult to interpret in the setting of focal brain injury or peripheral neuropathy.