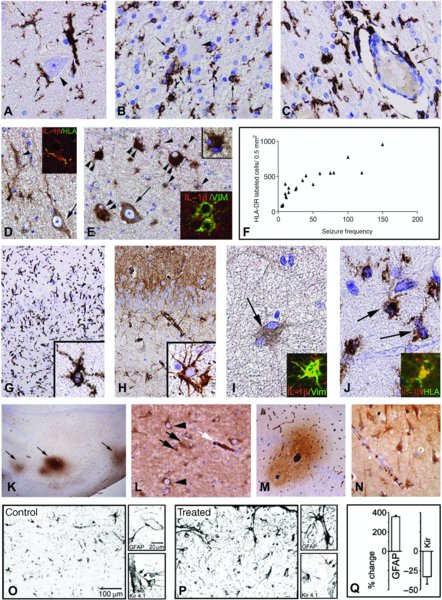
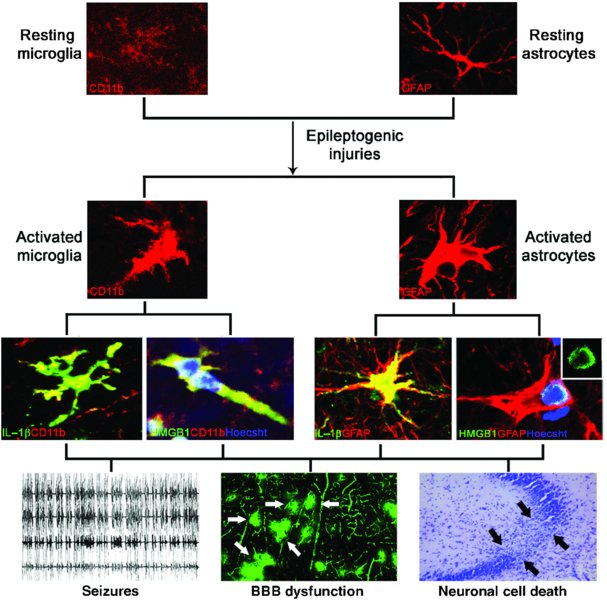
12 Jan Bauer1, Bethan Lang2, Sarosh R. Irani2 and Annamaria Vezzani3 1Department of Neuroimmunology, Center for Brain Research, Medical University of Vienna, Vienna, Austria 2Nuffield Department of Clinical Neurosciences, John Radcliffe Hospital, Oxford, UK 3Department of Neuroscience, Mario Negri Institute for Pharmacological Research, Milan, Italy Epilepsy is a brain disorder characterized by a predisposition to generate seizures and is frequently associated with cognitive and psychological sequelae. Seizures, the hallmarks of epilepsy, originate from synchronized aberrant firing of neuronal populations due to underlying hyper-excitability. Epilepsy occurs in about 1% of the general population; therefore, there are about 50 million people worldwide affected by this disease. Seizures can be caused by various aetiologies which include genetic factors, or they may be associated with focal damage to the brain. However, which factors actually trigger seizures remains unknown. Despite the availability of a wide range of antiepileptic drugs (AEDs) with predominant actions on voltage-dependent ion channels and classical neurotransmitter systems (Rogawski and Loscher, 2004), about one-third of affected individuals experience seizures which are not controlled by the available treatments. Therefore, there is an urgent medical need to develop new effective therapeutics. In addition, the available AEDs are predominantly symptomatic drugs that often do not interfere with the aetiopathogenic mechanisms underlying the onset of the disease or its progression (Pitkanen and Lukasiuk, 2011). Research for new therapies in epilepsy is reliant upon understanding the pivotal mechanisms of neuronal hyper-excitability. In a number of cases, this has led to the identification of immune or inflammatory processes in the epileptic brain in different forms of the disease, which may be involved in the pathogenesis of seizures. Clinical and experimental findings suggest that the activation of both the innate and adaptive immune systems, and the associated inflammatory processes, are involved in many forms of epilepsy. A role for inflammatory molecules in the generation of seizures has long been suspected since it was shown that selected anti-inflammatory treatments can control seizures in active and difficult-to-treat paediatric epilepsies. These treatments, in particular steroids and the use of intravenous immunoglobulins (IvIg) and adrenocorticotropic hormone (ACTH), are used in specific syndromes which are refractory to conventional AEDs (e.g. infantile spasms, continuous spikes and waves in sleep, and Rasmussen encephalitis (RE)). In recent years, a small but significant number of epileptic disorders have been associated with the presence of specific autoantibodies to neuronal receptors, ion channels or their accessory proteins in serum and cerebrospinal fluid (CSF). A pathogenic role for some of these autoantibodies is suspected based on clinical response to immunotherapy and other experimental observations. Indeed, the recent description of faciobrachial dystonic seizures (FBDS) has linked a clinically distinctive epilepsy with antibodies directed against the voltage-gated potassium channel (VGKC) complex protein LGI1 (Irani et al., 2008, 2011). Moreover, systemic and neurological autoimmune disorders have been associated with seizures or epilepsy in a small but significant number of cases (Palace and Lang, 2000; Vincent and Crino, 2011). These disorders are increasingly recognized and often treated successfully with immunotherapies. In addition, a careful assessment of inflammatory molecules and immune cells was performed in surgically resected brain tissue from patients with pharmaco-resistant epilepsies without an established or suspected autoimmune aetiology. In these common forms of epilepsy, the results revealed a significant component of the inflammatory response confined to resident brain cells (i.e. microglia, astrocytes and neurons) (Vezzani et al., 2011a; Aronica et al., 2012). Mechanistic insight into the role of these inflammatory molecules in the aetiopathogenesis of seizures demonstrated their prominent effects in inducing neuronal network hyper-excitability. First, these molecules can act as neuromodulators via stimulation of their cognate receptors expressed by neurons in diseased tissue. In addition, the effects of inflammatory molecules released by brain cells on blood–brain barrier (BBB) permeability function or astrocytic properties have been recently described to play important roles in tissue hyper-excitability (Vezzani et al., 2013). Proof-of-concept pharmacological evidence in experimental models of temporal lobe epilepsy (TLE) has identified anticonvulsive properties of anti-inflammatory drugs already used for other clinical indications. These drugs may be considered for prospective clinical trials for treating seizures not only in autoimmune forms but also in more common forms of epilepsy. The aim of this chapter is to outline the recent discoveries in this field by combining pre-clinical and clinical evidence of activation of the immune system and its functions in epilepsy. Mechanisms underlying the immune system’s effects in epileptic brain tissue will also be addressed. Finally, the current immunotherapies and the future challenges for new anti-inflammatory treatments in epilepsy will be discussed. As shown in animal models, the innate immune system may contribute directly to the induction of seizures (Vezzani et al., 2011a). Like other organs, the human central nervous system (CNS) contains cells (e.g. microglia and astrocytes) which after activation can secrete a large range of soluble inflammatory mediators, including interleukins (IL), chemokines, prostaglandins and complement components (Vezzani et al., 2011a). The activation of these cells and the concurrent production of inflammatory molecules have been shown not only in inflammatory and neurodegenerative diseases such as multiple sclerosis and Alzheimer’s disease (Link, 1998; McGeer and McGeer, 2002; Maccioni et al., 2009; Fernandez et al., 2010; Gandhi et al., 2010), but also in various epileptic disorders (Crespel et al., 2002; Boer et al., 2006; Choi et al., 2009; Wirenfeldt et al., 2009) (Figures 12.1 and 12.2). Thus, it is likely that innate immunity plays a significant role in epilepsies. One of the predominant players investigated in epilepsy research is IL1β (Figures 12.1 and 12.2). In a variety of human epileptic disorders (TLE, focal cortical dysplasia (FCD) type IIb, glioneuronal tumours and tuberous sclerosis), a moderate to strong up-regulation of IL1β expression is seen in (dysplastic) neurons, microglial cells and astrocytes (Ravizza et al., 2006, 2008; Iyer et al., 2010; Aronica and Crino, 2011). A link between IL1β and seizures is supported by the positive correlation between IL1β levels in surgically resected brain specimens from patients and the frequency of their epileptic seizures (Ravizza et al., 2006). The same positive correlation was found between the number of activated microglia, the frequency of seizures and the duration of epilepsy (Boer et al., 2006) (Figure 12.2). Interestingly, in FCD type I patients with intractable seizures, characterized by cortical dyslamination in the absence of obvious neuropathology or aberrant cells, only low IL1β immunoreactivity in some microglial cells was found (Iyer et al., 2010). In TLE, FCD type IIb and the glioneuronal tumours, the cells ((dysmorphic) neurons, balloon cells, microglia and astrocytes) that showed increased expression of IL1β also demonstrated up-regulation of its receptor IL1RI (Ravizza et al., 2006, 2008; Iyer et al., 2010). Notably, akin to experimental animals, the level of the anti-inflammatory molecule IL1 receptor antagonist (IL1ra) was moderate as compared to that of IL1β (Ravizza et al., 2006, 2008; Iyer et al., 2010), suggesting that key mechanisms controlling the extent and duration of inflammation are inefficient in brain tissue. Besides IL1β, IL6 has also been implicated in epilepsy. After tonic-clonic seizures in humans, increased levels of IL6 can be found in CSF (Peltola et al., 1998). Furthermore, elevated serum levels of IL6 are found in patients with refractory epilepsy (Hulkkonen et al., 2004). Conversely, a number of studies did not show differences in IL6 gene or protein expression in epileptic brains (van Gassen et al., 2008; Kan et al., 2012a,b). Thus, the role of IL6 in epilepsy requires further study. Figure 12.1 Neuropathology in different forms of human epilepsy. (A) Dystrophic neurons in the cortex of a focal cortical dysplasia (FCD) type IIb brain stained by antibodies against neurofilament (SMI32). M: meninges. Bar: 100 μm. (B) NeuN staining shows severe neuronal loss in the hippocampus of a temporal lobe epilepsy (TLE) with hippocampal sclerosis (HS) brain. Bar: 200 μm. (C) NeuN staining shows severe cortical degeneration with the presence of a few remaining neurons (arrow) in a Rasmussen encephalitis (RE) brain. Arrowheads indicate areas of vacuolization. M: meninges. Bar: 200 μm. (D–F) T lymphocytes in epilepsy. (D) Moderate numbers of CD3+ T cells in the cortex of FCD type IIb brain. Bar: 100 μm. (E) Moderate numbers of T cells in the hippocampus of a TLE–HS brain. Bar: 100 μm. (F) Infiltrate with CD3+ T cells in an RE brain. The inset shows the apposition of multiple cytotoxic (CD8+) T cells (blue) to neurons (brown: staining for NeuN). Bar: 100 μm. (G–I) Microglia activation in epilepsy. (G) Moderate up-regulation of CD68 in microglia in FCD type IIb brain. Bar: 50 μm. (H) Mild up-regulation of CD68 in microglia (arrowheads) is seen in a TLE–HS brain. Bar: 50 μm. Strong CD68 reactivity in microglia in an RE brain. Bar: 100 μm. (J and K) IL1β in an RE brain. (J) IL1β in a microglial nodule. Bar: 100 μm. (K) IL1β (arrowheads) in cells which probably are astrocytes. Bar: 100 μm. Figure 12.2 Panels A–E depict the distribution of microglia (A–D) and astrocytes (E) in focal cortical dysplasia and the production of IL1β in cellular elements within the dysplastic cortex (FCD type IIB) (D, E). Activated and ameboid HLA-DR-positive microglia (arrows) clustered around dysplastic neurons (arrowhead in A), balloon cells (arrows in B) and blood vessels (arrows in C) (adapted from Boer et al., 2006). Panel D depicts IL1β-positive microglia (arrowheads; colocalization between HLA-DR and IL1β in inset) clustered around IL1β-positive neurons (arrow). Panel E shows IL1β immunoreactivity in large dysplastic neurons (single arrow), astrocytes (single arrowheads and insets) and balloon cells (double arrowheads) within the dysplastic cortex (adapted from Ravizza et al., 2006). Panel F shows the significant positive correlation between HLA-DR cell counts and seizure frequency in patients (seizures/month) (adapted from Boer et al., 2006). Panels G–J depict the distribution of microglia (G) and astrocytes (H) and their IL1β production in the hippocampus of TLE patients with HS. Panel G shows strong HLA-DR immunoreactivity in activated microglia (inset); panel H shows increased vimentin (vim) immunoreactivity in reactive astrocytes (inset). Panels I and J depict IL1β immunoreactivity (arrows) in microglia and astrocytes, respectively (adapted from Ravizza et al., 2008). Panels K–N show serum protein extravasation in brain parenchyma in TLE with HS. Panel K depicts albumin immunoreactivity in the hippocampus around blood vessels (arrows); panel L shows albumin uptake in neurons (arrowheads) and in astrocytes (black arrows); white arrows depict albumin in a blood vessel (adapted from van Vliet et al., 2007). Panels M and N depict IgG extravasation around a blood vessel and its uptake by neurons, respectively (adapted from Rigau et al., 2007). Panels O–Q illustrate the reduction of Kir4.1 in activated GFAP-positive astrocytes in rat cortex exposed for 24 h to serum albumin (treated, P) versus control tissue (O) and their respective mRNA percentage changes compared to non-treated tissue (Q) (adapted from Ivens et al., 2007). Although most epilepsy research has focused on cytokines, other components of the innate immune system also have been investigated. Examples of these are endogenous ligands for the Toll-like receptors (TLRs) and receptors for advanced glycation end products (RAGE), such as high-mobility group box-1 (HMGB1). HMGB1 is a chromatin-bound factor that, following cell damage or neuronal hyper-excitability, translocates from the nucleus to the cytoplasm where it is released in the extracellular milieu and induces pro-inflammatory signals by stimulation of TLR4 or RAGE (Lotze and Tracey, 2005). In brain tissue from TLE or malformations of cortical development (MCD) patients, HMGB1–TLR4 signalling is up-regulated in resident brain cells, similar to observations in experimental models of epilepsy (Maroso et al., 2010). In addition, in MCD patients, up-regulation of TLR2 and RAGE has been found (Zurolo et al., 2011). A recent study performed with a multiplex immunoassay compared 39 immune modulators in TLE patients with (+) and without (−) hippocampal sclerosis (HS). The results showed up-regulation of 21 inflammatory mediators, including 10 cytokines and seven chemokines. Further analysis revealed that TLE+HS, TLE−HS and control groups could be distinguished based on unique expression patterns of cytokines and chemokines such as IL7, IL10, IL25 and CCL4, molecules which previously had not been implicated in TLE (Kan et al., 2012a). Also, complement factors are associated with seizure induction. In human TLE+HS, astroglial, microglial and neuronal up-regulation of complement factors C1q, C3c and C3d was observed, particularly within regions where neuronal cell loss occurs. The membrane attack protein complex (C5b–C9) was predominantly detected in activated microglial cells (Aronica et al., 2007). Microarray data on cortical tubers from patients with tuberous sclerosis complex revealed strong up-regulation (7 to 30 times) of complement mRNA (C factor I, C1R, C1s, C3 and C4a) (Boer et al., 2010). Also, in gangliomas, C1qa, C1qb, C1qc, C1r, C1s, C3, C4a and C7 showed more prominent expression than in control specimens. Finally, a role for innate immunity also can occur through activation of cyclooxygenase-2 (COX2), an inducible pro-inflammatory enzyme required for the synthesis of prostaglandins. COX2 was found to be up-regulated in neurons and astrocytes in human TLE+HS (Desjardins et al., 2003; Holtman et al., 2010). Experimental animal models of epilepsy have helped to address the crucial question of whether the complex and sustained inflammatory response, described in resected brain tissue from patients with refractory focal epilepsy, occurs as a cause of the underlying cellular pathology, as a consequence of seizures or both. In this context, experimental studies have demonstrated that recurrent seizures activate innate immunity in areas of seizure generation and generalization; in turn, the pre-existence of an inflammatory state in the brain induced by prototypical inflammatory molecules such as lipopolysaccaride or polyinosinic–polycytidylic acid (activating innate immunity via TLR4 and TLR3, respectively) can promote seizure activity and decrease seizure threshold (Riazi et al., 2010). The experimental studies were also instrumental in showing that the inflammatory molecules described in human epilepsy tissue play an active role in seizure mechanisms. Brain inflammation that is rapidly induced (<30 min) by an experimental epileptogenic injury (e.g. status epilepticus, traumatic brain injury, stroke or infection) outlasts the primary insult by days, and it is inefficiently opposed by endogenous anti-inflammatory mechanisms (Aronica and Gorter 2007; Bartfai et al., 2007; Vezzani et al., 2011a). Following a convulsive challenge provoked by administration of chemoconvulsants, but also after a brain injury which predisposes to epilepsy development, cytokines, prostaglandins, complement cascade and other downstream inflammatory molecules are induced, together with their cognate receptors, in neurons and in activated glial cells, as well as in endothelial cells of the BBB. These changes occur in the brain areas of focal injury, and in those affected by ongoing seizures. In the last decade, increasing knowledge of the mechanisms underlying seizures have shown that microglia and astrocytes significantly contribute to neuronal network dysfunctions (Fellin and Haydon 2005; Seifert et al., 2006; Halassa et al., 2007; Hanisch and Kettenmann, 2007). Recently, the crucial role played by pro-inflammatory molecules synthesized and released by activated glial cells in the mechanisms of seizures has gained increasing recognition (Aronica et al., 2012). It is now well established that, in addition to their involvement as effector molecules of immune activation following infections, pro-inflammatory molecules released by glia and neurons following various brain injuries may exert either direct or indirect neuromodulatory effects. The possibility that these molecules may contribute to aberrant neuronal excitability underlying seizure recurrence is a novel concept supported by clinical and experimental findings. The extent of brain inflammation in neurons and glia, as well as the activation of microglia in seizure-prone areas, positively correlates with the frequency of seizures and the severity of neuropathology both in animal models and in epilepsy patients (Boer et al., 2006; Ravizza et al., 2006; Iyer et al., 2010), showing a strict association between these events. Inflammatory molecules released by brain resident cells during seizures or following a brain injury may activate target cells (e.g. neurons, glia or endothelial cells of the BBB) to produce pathological effects. In particular, specific inflammatory pathways such as those activated by IL1β, transforming growth factor beta (TGFβ) or the complement cascade contribute not only to the recurrence of seizures but also to the precipitation of the first seizure. This was shown by pharmacological intervention to facilitate or block these pathways with concomitant proconvulsive or anticonvulsive effects, respectively. Pharmacological evidence that blockade of specific pro-inflammatory pathways affords anticonvulsive activity was supported by evidence showing decreased intrinsic seizure susceptibility in animals with genetic inactivation of the same inflammatory pathways (Vezzani et al., 2011a, 2013). Notably, experimental induction of a pro-inflammatory state in rat or mouse brain by activating TLR3 or TLR4 mediates a long-term increase in brain excitability, and favours seizure precipitation and excitotoxicity (Riazi et al., 2010). This study supports the concept that brain inflammation per se, before the onset of seizures, may contribute to excitability changes in brain tissue that are pivotal for epilepsy development (Bartfai et al., 2007; Pitkanen and Lukasiuk, 2011). Infection and fever, which are conditions associated with the activation of TLRs and rapid induction of pro-inflammatory molecules by cells of the innate immune system, are recognized precipitating factors of seizures (Singh et al., 2008). Therefore, common molecules and signalling pathways activated by either pathogens or brain injuries might contribute via converging innate immune mechanisms to the development of a chronic hyper-excitable neuronal network. In this context, the induction of IL1 receptor (IL1R)–TLR receptor signalling, which is pivotal for activation of innate immunity and inflammation following infections, is also induced by endogenous molecules released from injured cells (i.e. ‘danger signals’) such as HMGB1. This protein has proconvulsive properties (Maroso et al., 2010; Vezzani et al., 2011b). The activation of the IL1R–TLR signalling by IL1β and HMGB1 released following epileptogenic events occurs before seizure onset, but is also activated by recurrent seizures, thus establishing a pathologic vicious circle. Antagonism of the IL1–TLR pathway affords significant anticonvulsive activity and delays seizure onset in various animal models (Vezzani et al., 2011b; Aronica et al., 2012). Opening of the BBB has been suggested to be a crucial player in the generation of neuronal hyper-excitability (Figure 12.2). A positive correlation between the extent of BBB opening and the number of spontaneous seizures was shown in epileptic rats (van Vliet et al., 2007). Moreover, focal opening of the BBB in the rat neocortex resulted in the delayed development of paroxysmal hyper-synchronous activity (Seiffert et al., 2004; Ivens et al., 2007). This epileptogenic process was recapitulated by exposing the rat brain cortex to serum albumin. It was demonstrated that extravasation of serum albumin into the brain following local BBB breakdown activates a TGFβ receptor type 2 in astrocytes promoting local inflammation and astrocytic dysfunction (Cacheaux et al., 2009). In particular, albumin provokes transcriptional down-regulation of Kir4.1 potassium channels and of the glutamate transporter (Seiffert et al., 2004; Ivens et al., 2007; David et al., 2009; Friedman et al., 2009) in astroctyes (Figure 12.2). The resulting higher extracellular K+ and glutamate facilitate seizure precipitation and recurrence. Notably, inflammatory molecules produced and released by perivascular glia have a prominent role in BBB breakdown by provoking the down-regulation of tight junctions on microvascular endothelial cells (Morin-Brureau et al., 2011; Librizzi et al., 2012). To address the role of specific inflammatory molecules in epilepsy development after a primary brain injury, non-steroidal anti-inflammatory drugs, immunosuppressant molecules, and drugs inhibiting glia activation were administered to experimental animals after induction of status epilepticus. These post-injury treatments decreased the recurrence of spontaneous seizures in epileptic rats and ameliorated the associated neuropathology without interfering with the onset of the disease (Ravizza et al., 2011). Cognitive dysfunction and depression-like behaviours were also reduced in some instances (Mazarati et al., 2010, 2011; Riazi et al., 2010; Pineda et al., 2012). In summary, increasing experimental evidence suggests that pharmacological targeting of brain-borne inflammatory molecules may be a promising strategy for seizure control and possibly to modify the disease course after a challenging event (Figure 12.3). Additional studies are required to further characterize the detrimental inflammatory signals induced by brain injury and seizures, and to understand which treatments given before epilepsy onset in individuals at risk could potentially arrest the disease progression. Figure 12.3 Functional consequences of the production of inflammatory mediators by microglia and astrocytes. Epileptogenic brain injuries activate glial cells which in turn release inflammatory mediators such as IL1β and HMGB1, triggering brain inflammation. This event leads to changes in brain physiology provoking neuronal hyper-excitability, blood–brain barrier dysfunction and cell damage that contribute to lower seizure threshold and trigger epileptogenesis. There are two major direct mechanisms by which the adaptive immune system can play a role in epilepsy or epileptic disorders: antibody-mediated mechanisms and T cell cytotoxicity. Notably, immunohistochemical investigations of brain tissue from drug-resistant focal-onset epilepsies demonstrated albumin and IgG extravasation in the brain parenchyma, indicating BBB damage. These macromolecules have been found both extracellularly and within glial cells and neurons, suggesting a possible role in cell dysfunction and immune activation (Rigau et al., 2007; van Vliet et al., 2007; Ravizza et al., 2008). Infiltration of immunoglobulins and T lymphocytes in the brain can be found in a variety of encephalitis, including Rasmussen’s encephalitis (RE), a variety of viral encephalitis and a group of encephalitides which are defined by the presence of specific neural antibodies in serum and/or CSF. The latter, so-called antibody-associated encephalitis, is most commonly non-paraneoplastic but can also be paraneoplastic. For a long time, it was thought that antibody-mediated mechanisms played an important role in Rasmussen’s encephalitis (RE). RE can be modelled in rabbits following immunization with recombinant glutamate receptor type-3 (GluR3). Animals developed severe seizures and had inflammatory histopathological changes in their brains, similar to those found in patients with RE. Furthermore, antibodies to GluR3 were detected in the sera of some RE patients, and plasma exchange in one child significantly reduced the serum titres of GluR3 autoantibodies, decreased seizure frequency and improved neurologic function (Rogers et al., 1994). However, the pathological significance of anti-GluR3 antibodies diminished when it was shown that these antibodies were only infrequently found in RE or intractable epilepsies (Wiendl et al., 2001; Mantegazza et al., 2002), and that few patients improved clinically after plasmapheresis (Andrews et al., 1996; Granata et al.
Immune Responses in the CNS in Epilepsy
Introduction
Innate immunity in epilepsy
Human epilepsy

Experimental studies
Seizure-induced brain inflammation
Pre-existing inflammation and seizure susceptibility
Blood–brain barrier and inflammation
Anti-inflammatory drugs and epileptogenesis
Adaptive immunity in epilepsy
Human epilepsy
Rasmussen’s encephalitis
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel







Full access? Get Clinical Tree


