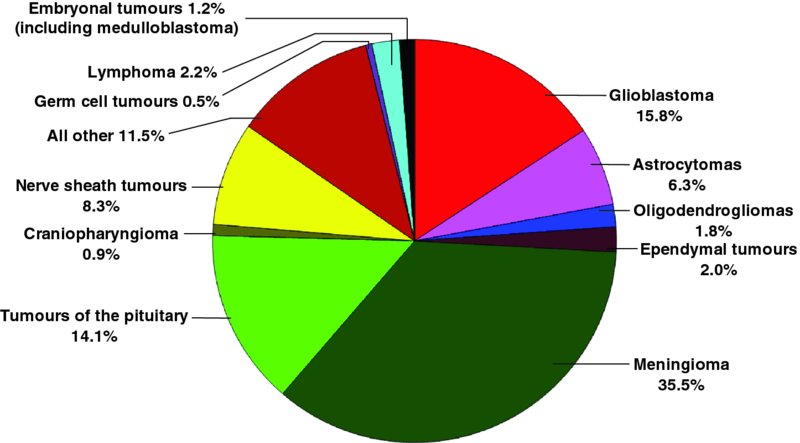
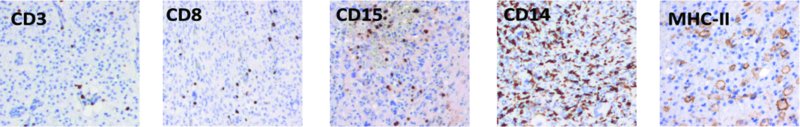
15 Paul Gielen1, Paul van der Valk2 and Pieter Wesseling3 1Department of Tumor Immunology, Nijmegen Centre for Molecular Life Sciences (NCMLS), Radboud University Medical Centre, Nijmegen, the Netherlands 2Department of Pathology, VU University Medical Center, Amsterdam, the Netherlands 3Department of Pathology, Radboud University Medical Centre, Nijmegen, and VU University Medical Center, Amsterdam, the Netherlands Compared to tumours elsewhere in the human body, the diversity and complexity of CNS tumours have been described as being unrivalled. Based on systematic histopathological analysis for more than a century, a World Health Organization (WHO) scheme of CNS tumours has emerged that is now accepted worldwide as the gold standard for classification of these neoplasms (Louis et al., 2007). An important distinction in the spectrum of CNS tumours is that of primary (i.e. arising from the CNS or its coverings) versus secondary neoplasms (i.e. metastases in or around the CNS from malignant tumours elsewhere in the body). Although primary tumours are much less common than CNS metastases of, for example, lung and breast cancer, they are on the top of the list of ‘average number of years of life lost’ by cancer (Burnet et al., 2005). This can at least partly be explained by two important facts: (i) the most frequent primary brain tumour is glioma, and most gliomas show very extensive, diffuse infiltrative growth in the surrounding CNS parenchyma, precluding curative therapy (Claes et al., 2007); and (ii) CNS tumours are relatively frequent in children, and many of those are highly aggressive. Although CNS tumours have been, up till now, classified based on their histopathological characteristics, during the last decade insight into the molecular aberrations underlying their oncogenesis has increased in a revolutionary way. It is increasingly clear that a combination of morphological and molecular characteristics will soon allow for a much more robust and clinically meaningful ‘taxonomy’ (typing and grading) of CNS tumours, especially of gliomas and embryonal tumours. Also, the molecular underpinnings provide information on the pathways that might be targeted with different therapeutic compounds. Increased knowledge about the genetic and molecular aberrations underlying oncogenesis has accumulated in a landmark article summarizing the tasks that a cancer cell generally needs to accomplish to successfully survive and grow as a tumour: sustain proliferative signalling, evade growth suppressors, resist cell death, enable replicative immortality, induce angiogenesis and activate invasion and metastasis (Hanahan and Weinberg, 2000). More recently, conceptual progress resulted in the addition of two hallmarks: reprogramming of energy metabolism and evading of immune destruction. Furthermore, it was acknowledged that genome instability in tumour cells and inflammation are characteristics facilitating or even enabling the hallmarks just listed (Hanahan and Weinberg, 2011). The most frequent CNS tumours will now be briefly introduced. Figure 15.1 compares the frequency of primary tumours of the CNS. Figure 15.1 Relative frequency of primary tumours of the CNS in 311,202 patients from 2005 to 2009 as registered by the Central Brain Tumor Registry of the United States (CBTRUS) (see Dolecek et al., 2012). Gliomas are considered to originate from glial cells or their precursors and may show astrocytic, oligodendroglial or ependymal differentiation or a combination thereof. In adult patients, most gliomas belong to the category of ‘diffuse gliomas’ (i.e. glial tumours showing extensive, diffuse, infiltrative growth in the surrounding brain parenchyma). The diffuse gliomas are astrocytic, oligodendroglial or mixed oligoastrocytic in nature. Based on histopathological parameters such as marked mitotic activity, a peculiar form of angiogenesis (‘florid’ or even ‘glomeruloid’ microvascular proliferation) and/or necrosis, a malignancy grade can be assigned to these tumours. The least malignant diffuse glioma is designated as WHO grade II, and the most malignant (and unfortunately also the most frequent) astrocytic tumour is traditionally coined as glioblastoma (WHO grade IV). In between these is a category of anaplastic (WHO grade III) diffuse gliomas. Both low-grade (WHO grade II) and high-grade (WHO grade III and IV) diffuse gliomas occur in all age groups, but glioblastomas are relatively frequent in patients older than 50 years of age, whereas young adults are more often diagnosed with a diffuse low-grade glioma. Unfortunately, these latter tumours have a strong tendency to progress to more malignant neoplasms in the course of years. Important other groups of gliomas include the ependymomas and ‘variant astrocytic gliomas’. These ‘other’ gliomas tend to show more circumscribed growth and to occur much more frequently in the paediatric and young adult age group. Although ependymomas can be low- or high-grade malignant, most ‘variant astrocytic gliomas’ are low grade (WHO grade I or II) and, in contrast to diffuse gliomas, do not show progression to a more aggressive lesion in the course of time. By far the most frequent ‘variant glioma’ in the paediatric age group is pilocytic astrocytoma, a WHO grade I lesion that most frequently occurs in the cerebellum, the hypothalamic region and/or the optic nerve. About 5 years ago it was demonstrated that WHO grade II and III diffuse gliomas in adult patients often carry an isocitrate dehydrogenase 1 (IDH1) or (less frequently) IDH2 mutation. Earlier, complete co-deletion of the short arm of chromosome 1 and of the long arm of chromosome 19 (complete 1p/19q co-deletion) was reported as a characteristic molecular aberration for oligodendroglial tumours, whereas high copy amplification of the epidermal growth factor receptor (EGFR) and the presence of its variant III (EGFRvIII) are typically found in glioblastomas (Riemenschneider et al., 2010). Diffuse gliomas in the paediatric age group have a different molecular background, with relatively frequent mutations in Histon-H3 or the ATRX (alpha thalassemia–mental retardation syndrome X-linked) gene (Sturm et al., 2012). Pilocytic astrocytomas often harbour aberrations related to BRAF (the BRAF V600E mutation or KIAA1549–BRAF fusion gene) (Pfister et al., 2010). Using gene expression data from the Cancer Genome Atlas Project, gliomas can be divided into four subgroups: classical, neural, proneural and mesenchymal. These subtypes are associated with aberrations of EGFR, neurofibromin-1 (NF1), platelet-derived growth factor receptor alpha (PDGFRA) and IHD1. Furthermore, a link was described with response to therapy, with aggressive treatment being the most effective for the classical subtype and having a more limited effect on tumours of the proneural subtype (Verhaak et al., 2010). Tumours originating from meningothelial cells and arachnoid cap cells in the (lepto)meninges are traditionally designated as meningiomas. The vast majority of these tumours are benign (WHO grade I) and show a compact, expansive growth pattern, allowing for relatively easy surgical removal (or highly focussed irradiation in cases where the tumour cannot be adequately removed by surgery). Based on increased mitotic activity and/or a number of other microscopic features (including necrosis, high cellularity and patternless growth) a small subset of meningiomas is graded as WHO grade II (‘atypical’; up to 8% of meningiomas) or even as WHO grade III (‘anaplastic’ or malignant; 1–2% of meningiomas). Also, meningiomas showing infiltrative growth in the brain parenchyma are known to be associated with a substantially higher recurrence rate, comparable to atypical meningiomas, and are therefore also graded as WHO grade II. CNS tumours characterized by high cellularity with small, (apparently) poorly differentiated, highly proliferative tumour cells are grouped together as the high-grade malignant (WHO grade IV) category of ‘embryonal tumours’. Many of these tumours show signs of neuronal differentiation at the immunohistochemical level and can be designated as primitive neuro-ectodermal tumours (PNETs). PNETs are rare in adult patients but relatively frequent in the paediatric age group (about 15% of CNS tumours in children aged 0–14 years) (Dolecek et al., 2012). By far, the most common representative in this category is medulloblastoma, a neoplasm that by definition originates in the posterior fossa/cerebellum. Morphologically, medulloblastomas are subtyped as classic, desmoplastic/nodular, extremely nodular, and large cell/anaplastic. The desmoplastic and extremely nodular variants are associated with a significantly better prognosis, whereas the large-cell or anaplastic subtype shows an even more aggressive behaviour compared to classic medulloblastomas. Recently, molecular characteristics were elucidated that not only provide more robust information on the clinical behaviour that is to be expected, but also may help to design and implement more effective, molecularly targeted therapies (Northcott et al., 2012). Other relatively frequent embryonal tumours of the CNS are supratentorial PNETs, pineoblastomas and atypical teratoid–rhabdoid tumours (AT/RTs). Supratentorial PNETs and pineoblastomas are often morphologically and immunohistochemically indistinguishable from classic medulloblastomas, and it is then its site of origin that determines the precise name that is given to the lesion. It is increasingly clear, however, that these tumours indeed differ not only in their location but also in their molecular underpinnings. AT/RTs are relatively recently defined as a separate group of embryonal tumours, with typically ‘rhabdoid features’ (globular eosinophilic condensations) in the cytoplasm of (part of) the tumour cells. Of note, microscopic delineation of PNETs from AT/RTs can be difficult, especially in cases in which the rhabdoid features are not that prominent. Here, too, molecular markers are very helpful as most AT/RTs show loss of function of the SMARCB1 tumour suppressor gene (SWI–SNF (switch/sucrose nonfermentable) related, matrix-associated, actin-dependent regulator of chromatin subfamily B member 1; also called INI1) or, less frequently, of the SMARCA4 gene (Hasselblatt et al., 2011). Relatively frequent other primary tumours of the CNS are pituitary adenomas (generally benign, but diverse with regard to hormonal production), schwannomas (benign, originating around the CNS from the roots of the cranial nerves (relatively frequently in the cerebellopontine angle) or from the nerve roots in the spinal canal) and primary CNS lymphomas (over 95% of these being high-grade malignant, diffuse large B cell lymphomas and relatively frequently occurring in immune-compromised patients). Less frequent neoplasms include primary germ cell tumours of the CNS, a very heterogeneous group of variable malignancy (with germinomas, embryonal carcinomas, choriocarcinomas and yolk sac tumours being high-grade malignant, and mature teratomas being benign), and haemangioblastoma (benign, extremely well-vascularized and often partly cystic neoplasms that occur in the context of Von Hippel–Lindau disease in about one-quarter of patients with a haemangioblastoma). Secondary or metastatic tumours in and around the CNS are the most frequent CNS neoplasms. Some cancers show a significantly higher propensity to form CNS metastases (particularly carcinoma of the lung, breast, kidney and melanoma) than others (e.g. colorectal and prostate cancer). Also, the metastatic lesions may be single (and thereby more easily amenable to local surgical therapy and/or intense irradiation) or multiple, and metastases may occur in different compartments of the CNS (intraparenchymal, leptomeningeal and intraventricular). A very important aspect in clinical neuro-oncology is the fact that the CNS microvasculature normally forms a blood–brain barrier (BBB). This barrier is elicited by the interaction between the microvascular endothelial cells and the perivascular astrocytic endfeet. The barrier function is established by a combination of tight junctions between the endothelial cells, only limited trans-endothelial transport via pinocytotic vesicles and the presence of transporter molecules on the endothelial cells that are specialized in keeping out compounds that are harmful for the brain parenchyma (Daneman, 2012). Understandably, this barrier function contributes to reduced efficacy of chemotherapeutic compounds for CNS tumours. Furthermore, because of the presence of the BBB, the CNS has for many years been considered as an immune-privileged site (De Micco, 1989; Mitchell et al., 2008) (see Chapter 1). When performing T1-weighted magnetic resonance imaging (MRI) after intravenous injection of the paramagnetic contrast agent gadolinium–diethylenetriaminepentacetate (gadolinium–DTPA, an agent that passes the blood vessel wall only when the BBB is defective), contrast enhancement of a CNS tumour unequivocally means loss of the barrier function in the enhancing area. In diffuse low-grade gliomas, contrast enhancement is generally absent, indicating that the infiltrating glioma cells in the brain parenchyma leave the BBB relatively unaffected. In many other CNS tumours, however, the BBB function is incomplete or even lost (Claes et al., 2007). This is true not only for high-grade malignant tumours located in the brain parenchyma (‘intra-axially’) such as high-grade gliomas and metastases, but also for benign and WHO grade I neoplasms such as pilocytic astrocytomas and haemangioblastomas. Meningiomas generally show intense contrast enhancement but these neoplasms are located outside the brain parenchyma (‘extra-axially’). In a subset of meningioma patients, the surrounding brain tissue shows oedema but without contrast enhancement. Based on examination of haematoxylin-and-eosin (H&E)-stained sections, neuropathologists are well aware of the fact that striking inflammatory infiltrates may occur in CNS tumours with a highly variable contribution of lymphocytes, plasma cells, macrophages and neutrophilic and/or eosinophilic granulocytes. Generally, the presence or absence of such an infiltrate does not have an impact on the diagnosis. Rather, the histopathological diagnosis is based on the characteristics of the tumour cells themselves and on the results of some other tumour–microenvironment interactions such as infiltrative growth, necrosis and microvascular changes, whereas the inflammatory infiltrate is often considered as something that ‘just happens’. This is especially true in cases where inflammatory cells can be explained as a logical consequence of, for example, hypoxia and necrosis (macrophages). Microglial cells, the resident, antigen-presenting cells of the CNS, are inconspicuous in most routinely performed histochemical stainings. Some CNS tumours are well known for the occurrence of a particularly prominent lymphocytic infiltration. Examples are diffuse astrocytic tumours with gemistocytic change (i.e. ‘ballooning’ of the cytoplasm of the astrocytic tumour cells because of accumulation of glial fibrillary acidic protein (GFAP) filaments) and ‘variant’ gliomas such as pleomorphopic xanthoastrocytoma and ganglioglioma that often show dispersed, prominent perivascular cuffs of small lymphocytes. Furthermore, germinoma (the most frequent primary germ cell tumour of the CNS, occurring especially in the pineal gland of adolescent boys) classically shows a combination of clusters of large tumour cells and dense infiltrates of small lymphocytes. Thereby, germinomas look exactly the same as their counterparts originating in the testis (seminoma) and ovary (dysgerminoma). Like lymphomas elsewhere in the body, primary CNS lymphomas are composed of a mixture of lymphoid tumour cells (in over 95% of the cases, large and immature B cells) and non-neoplastic inflammatory cells, especially lymphocytes. In Langerhans cell histiocytosis (LCH) (a rare ‘haematopoetic tumour’ that most often occurs in children and young adults), remarkable accumulation of eosinophilic granulocytes is often seen. The presence of prominent infiltrates of lymphocytes (or eosinophilic granulocytes) may thus guide the neuropathologist towards the correct diagnosis and occasionally even allows for the diagnosis of a specific tumour subtype: in the lymphoplasmacyte-rich subtype of meningioma, the accumulation of lymphocytes and plasma cells can be so intense and widespread that the meningothelial tumour cells are difficult to identify (Louis et al., 2007). Soon after immunohistochemical staining became available, it was clear that the number of inflammatory cells in CNS tumours was often much higher than had been recognized in H&E-stained sections (Figure 15.2). For instance, in an immunohistochemical study using antibodies for microglial cells and macrophages, large numbers of these cells could be demonstrated in gliomas (Roggendorf et al., 1996). Furthermore, the inflammatory cells in CNS tumours could be further characterized, and differences between different (sub)types of tumours could be investigated. For example, in a study of glioblastomas and pilocytic astrocytomas, glioblastomas exhibited significantly higher perivascular cytotoxic CD8+ T cell infiltration, as well as more perivascular and intratumoural CD56+ natural killer (NK) cells and CD68+ macrophages. The CD3, CD20 and leukocyte common antigen (LCA)-positive cells did not differ between glioblastoma and pilocytic astrocytoma (Yang et al., 2011). The same group reported that a relatively high CD8+ T cell infiltrate in newly diagnosed glioblastoma was associated with prolonged survival (Yang et al., 2010b). In another study of glioblastoma patients, a moderate but not significant inverse association between regulatory T cells (Tregs) and survival was noted (Jacobs et al., 2010). Figure 15.2 Examples of immunohistochemical detection of immune cells in glioblastoma; histological sections of formalin-fixed, paraffin-embedded tumour tissue showing the presence of T cells (CD3), cytotoxic T cells (CD8), granulocytic cells (CD15) and microglia and macrophages (CD14, MHC-II).
Immune Responses to Tumours in the CNS
Tumours of the CNS
Primary tumours of the CNS
Gliomas
Meningiomas
Embryonal tumours
Other primary tumours
Secondary tumours
Blood–brain barrier in CNS tumours
The inflammatory infiltrate in CNS tumours
A traditional neuropathologist’s perspective
Revelations by immunochemistry
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


