Fig. 1
The effect of various concentrations of E2 on NO production in bEnd.3 cells after 2 h of treatment. Nitrite (NO2; a metabolite) was measured as a marker for NO production. There is a significant increase with all three E2 concentrations (0.1, 10, and 1,000 nM) when compared with the control cell population (p < 0.05). However, there is no significant difference within the applied E2 concentrations. Addition of ICI (100 nM, inhibitor of ER) as well as 1400W (10 μM, antagonist of NOS) significantly inhibited the E2-induced NO production (p < 0.05)
eNOS expression was detectable in cell lysates (Fig. 2). E2 induced increases in eNOS protein levels at concentrations of 1,000 nM, suggesting that E2 controls NO production by transcriptional activation of eNOS. There was no effect on eNOS protein expression at 10 nM, but increased levels of NO2 were detected, implying that there is posttranscriptional activation of eNOS at this concentration. This fact is supported by elevated levels of phosphorylated eNOS (eNOSp) after E2 treatment (Fig. 3).
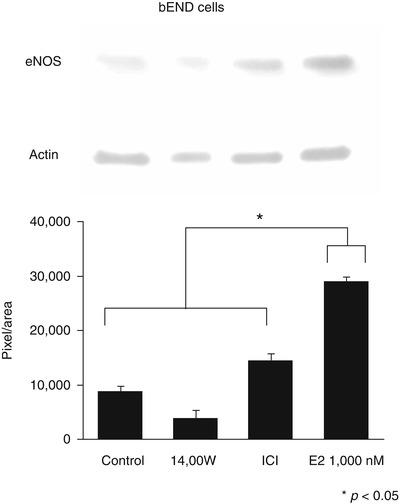
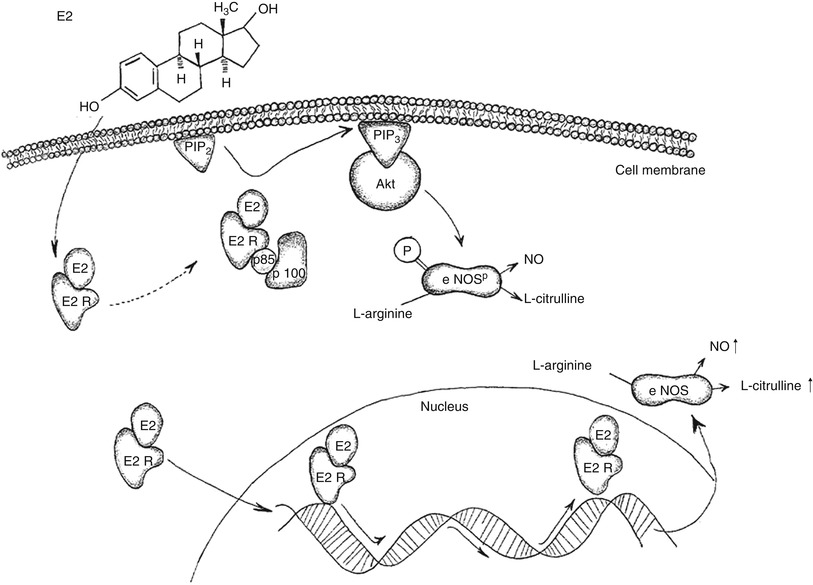
Fig. 2
(Upper part) Western blot analysis of eNOS expression in control bEnd.3 cells, bEnd.3 cells treated with 1400W, ICI, and E2 concentrations at 1,000 nM after 2 h of treatment. (Lower part) Quantitative Western blot gel analysis. The y-axis shows mean (n = 3 measurements) pixel per area calculated with NIH Image J software. Error bars are standard deviations (p < 0.05)
Fig. 3
Schematic illustration of postulated E2 effect in bEnd.3 cells. E2 binds to its receptor E2R. One pathway to increases NO production is by activation of phosphatidylinositol 3-OH kinase (PI3K). PI3K is a heterodimeric phosphoinositide kinase consisting of a regulatory (p85) and catalytic subunit (p100). E2R binds to the catalytic subunit of PI3K and leads to the production of phosphatidylinositol 3-phosphate (PIP3), which, in turn, activates the serine–threonine protein kinase B (Akt). Akt leads to a rapid activation an eNOS, accomplished through phosphorylation at serine1177. Another pathway of E2 is through directly enhancing gene expression of eNOS. This, in turn, enhances intracellular protein levels of eNOS in bEnd.3 and increases NO production
Discussion
This study indicates that E2 induces NO level increases in cerebral and peripheral endothelial cells in vitro via eNOS activation and through E2-receptor mediated mechanisms. This is presumably mediated through activation of eNOS by Akt-dependent phosphorylation and via transcriptional activation of eNOS.
NOS Subtypes and E2 Receptors
Among the three different NOS types, there are two consecutive Ca2+/calmodulin-dependent isoforms; namely neuronal NOS (nNOS) and endothelial NOS (eNOS). nNOS is widely distributed in the central nervous system, where NO production occurs via activation of nNOS in neural cells [21, 23]. eNOS is distributed throughout the cardiovascular system, including cerebrovascular tissue, where its activity leads to NO production in endothelial cells [16, 19]. Shear stress to blood vessels [5] and E2 [3] are two of the few agonists that can activate eNOS in a Ca2+-independent manner. Phosphorylation of human eNOS at Serine1177 results in enhanced eNOS activity and increased NO release [5, 9]. The inducible NOS (iNOS) is a Ca2+/calmodulin-independent isoform and is mainly expressed in activated immune cells, including microglia of the brain [17].
Currently, three different types of E2 receptors (ER) are known, namely ERα, ERβ, and a G-protein-coupled receptor, GPR30/GPER. ERα and ERβ act as ligand-dependent nuclear transcriptional factors; ERα also possesses ligand-independent activity. These two nuclear transcriptional factor ER receptors share a high homology in the regions that bind DNA and can form heterodimers. All three receptors are found in the brain and play important roles in neuroprotection, though the role of ERα appears most prominent [22]. Classically, the sex hormone estrogen exerts its effects by modifying gene expression [2, 12]. However, it has been shown that ERα mediates rapid, nongenomic effects in human endothelial cells by activating eNOS, suggestive of a process that does not involve the nuclear effects of the hormone [4]. These effects, mediated by estrogens, are too rapid to be elicited by protein synthesis and are not blocked by inhibitors of transcription. This novel activity has promoted the search for alternative signaling mechanisms and led to the term “nongenomic” actions of E2 or membrane-initiated steroid signaling (MISS). Hynes et al. were able to show that E2 leads to eNOS activation via the phosphatidylinositol 3-OH kinase (PI3K)– serine–threonine protein kinase B (Akt) pathway [11]. Further, it was shown that ERα couples with the regulatory subunit of the lipid kinase, PI3K, activating the catalytic subunit and increasing intracellular production of phosphoinositides (PI) [24]. PI3K is a heterodimeric phosphoinositide kinase consisting of a regulatory and catalytic subunit [1]. PI3K transfers the signaling cascade to intracellular protein kinases by phosphorylation of phosphatidylinositol at the D-3 position, which, in turn, acts as a lipid-mediated second messenger. Akt is one of the principle targets of this cascade. Many of the downstream effects of PI3K are mediated by activated Akt, including rapid eNOS activation, accomplished through eNOS phosphorylation at serine1177 [5, 9].
bEnd.3 cells express both isoforms (ERα and ERβ) of the estrogen receptor [18], therefore we chose Fulvestrant (ICI) to completely block the ERs, which, in turn, inhibited E2-triggered NO production. E2 led to a significant increase of NO2 in bEnd.3 and HUVECs by increasing eNOS protein levels and also increased levels of eNOSp. Because the administration of a potent eNOS inhibitor (1400W) inhibited the increase of NO2 levels in bEnd.3 cells, we assume that E2 has a direct effect on eNOS in these cells. It has been shown that E2 increases the expression of NOS [26]. Aside from the increased protein expression induced by E2 in both cell lines in our study, we postulate that increased levels of eNOSp are activated by Akt-mediated pathways. Both cell lines revealed similar molecular effects after estrogen treatment, suggesting that brain and peripheral endothelial responses to E2 have similar outcomes.
Relation to Vasospasm and DIND After SAH
Cerebral vasospasm itself as part of a complex pathophysiology has multiple causal factors. One such factor is deprivation of nitric oxide (NO) in the vicinity of brain vessels as a result of nitric oxide synthase (NOS) dysfunction [20] and scavenging of NO by deoxyhemoglobin [15]. Enhanced perivascular NO concentration has been shown to prevent or reverse vasospasm [7, 8]. Administration of NO donors may lead to several systemic adverse effects, including development of drug tolerance, systemic hypotension, rebound phenomenon, and brain edema [6]. To avoid these side effects, direct activation of eNOS, and thus increasing endogenous NO levels, has emerged as a promising research direction. E2 has been shown to reduce mortality and secondary ischemic damage [27] and prevent cerebral vasospasm following SAH in animal models [13]. The results of our in vitro study demonstrate the direct effect of E2 in brain endothelial cells by activating eNOS and raising NO levels by transcriptional and posttranscriptional mechanisms, warranting further confirmation in future in vivo studies.
Related posts:
 Role of Erythropoietin in Aneurysmal Subarachnoid Haemorrhage: From Bench to Bedside
Modulation of Spreading Depolarizations
Factors of Temporary Arterial Occlusion During Anterior Communicating Artery Aneurysm Repair
Impairment in Aneurysmal Subarachnoid Hemorrhage Patients with Delayed Cerebral Infarction: Prevalence and Pattern
Role of Erythropoietin in Aneurysmal Subarachnoid Haemorrhage: From Bench to Bedside
Modulation of Spreading Depolarizations
Factors of Temporary Arterial Occlusion During Anterior Communicating Artery Aneurysm Repair
Impairment in Aneurysmal Subarachnoid Hemorrhage Patients with Delayed Cerebral Infarction: Prevalence and Pattern
 Intracranial Arterial Pressure Monitoring During Endovascular Cerebral Aneurysms Embolization for Cerebral Perfusion Evaluation
Intracranial Arterial Pressure Monitoring During Endovascular Cerebral Aneurysms Embolization for Cerebral Perfusion Evaluation
 Secondary Injury After Subarachnoid Hemorrhage in Light of Multimodal Advanced Neuroimaging, Intracranial and Transcranial Neuromonitoring: Beyond Vasospasm
Secondary Injury After Subarachnoid Hemorrhage in Light of Multimodal Advanced Neuroimaging, Intracranial and Transcranial Neuromonitoring: Beyond Vasospasm
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


