A phase-specific neuroimmune model of clinical depression with remission.
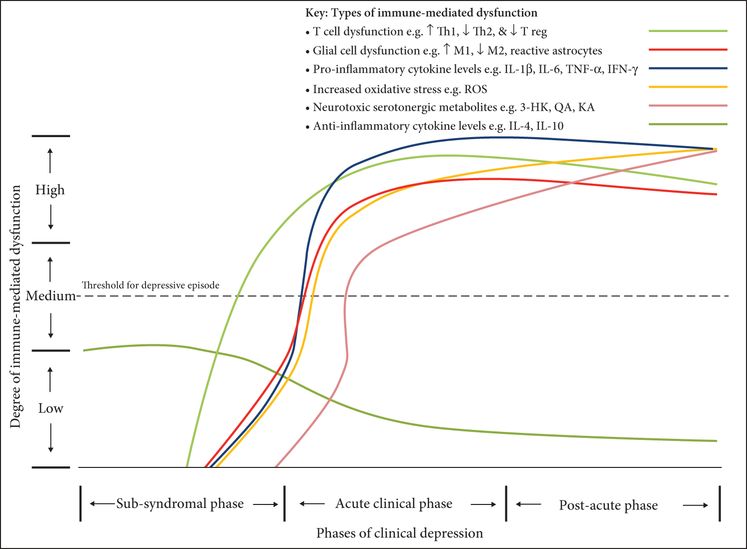
A phase-specific neuroimmune model of clinical depression: chronic major depressive episode with progressive depressive features and cognitive dysfunction.
The sub-syndromal phase encompasses factors relevant to depression prior to the onset of clinically significant symptoms. These include sub-threshold symptoms and risk factors for the development of depression. In this phase, a proportion of patients may not go on to develop a depressive episode, with their sub-threshold symptoms reducing shortly thereafter. Some patients may go on to develop a major depressive episode.
The acute clinical phase of the model encompasses the initiation and main duration of the clinically significant part of the depressive episode, when there is maximal symptomatology. Depressive symptoms may be mild, moderate, or severe and may include melancholic, atypical, psychotic, and non-melancholic symptomatology. This phase can be the first episode for an individual and may form a single episode with no lifetime relapse; however, this phase may instead connote a recurrence of depression with a prior period of total or partial remission. Finally, this stage may represent a progressive recurrence of depression whereby each episode can be associated with more severe symptomatology and a longer duration than the previous episode (Moylan et al., 2013).
The post-acute phase encompasses a period when depressive symptoms are generally improving. During this period there can be full recovery whereby the symptomatology of the affected individual is the same as before the depressive episode; alternatively, there can be partial recovery. The final possibility is there is no recovery and the affected individual’s symptoms become chronic and neurocognitive decline is progressive.
Molecular mechanism of inflammation impacting on the symptom domains of cognitive function and emotion regulation in depression
The effects of cytokines on the response of an individual to biologically relevant stimuli have been studied extensively in the past few decades. The most important results in this category are the ability of cytokines to affect cognitive performance and emotional processing and to induce sickness behavior in mammals (Anisman, Kokkinidis, & Merali, 2002), which includes anhedonia, feelings of depression and guilt, decreased sexual behavior, and a decreased appetite. Centrally administered IL-1β has been shown to inhibit food intake and social behavior (Connor, Song, Leonard, Merali, & Anisman, 1998; Brebner, Hayley, Zacharko, Merali, & Anisman, 2000; Nadjar, Bluthe, May, Dantzer, & Parnet, 2005). Centrally administered TNF-α demonstrated equal results and also increased lying down (compared with standing) (Brebner et al., 2000; Sakumoto, Kasuya, Komatsu, & Akita, 2003). Other cytokines, such as IFN (Hurlock, 2001) and IL-6 (Connor et al., 1998), have also been shown to affect the response to biologically relevant behavior. However, none of these studies have focused especially on the role of the amygdala in these processes. Since the amygdala is involved in the response to biologically relevant stimuli (Dougherty & Rauch, 2001), and cytokines have been shown to affect the amygdala (Kaur & Salm, 2008), it is to be expected that cytokines may act on this response by acting on the amygdala. However, the available evidence so far has not proven this hypothesis. Until now, cytokines have been shown to act on the response to biologically relevant stimuli mainly by acting on other brain regions, either by affecting the HPA axis (Ericsson, Kovacs, & Sawchenko, 1994) or by up-regulation of several neurotransmitters (Dunn, 2006). Though the role of the amygdala in the response to biologically relevant stimuli as a result of administration of cytokines is not ruled out, further research on these processes is required.
Molecular mechanisms underlying cytokine effects on cognition
It is now believed that cytokine-mediated pathophysiological processes underlie the cognitive impairments associated with several neuropsychiatric diseases including depression, making cytokines ideal targets for therapeutic intervention (Reichenberg et al., 2001; Wilson, Finch, & Cohen, 2002; Tobinick, 2007; Tweedie, Sambamurti, & Greig, 2007). Despite these pathological implications (Bitsch et al., 2000; He, Zong, Wu, Wei, & Xiang, 2007b), cytokines have also been shown to exert physiological (Blatteis, 1990; Sei, Vitkovic, & Yokoyama, 1995; Vitkovic, Bockaert, & Jacque, 2000a) and even neuroprotective (Schwartz et al., 1991, 1994) functions.
Recent findings suggest that in healthy elderly humans, cytokines, in particular IL-8, under physiological conditions, are possibly involved in cognitive processes such as memory, perceptual speed, and motor function (Baune et al., 2008a). Moreover, the associations between genetic variants of cytokines (IL-1b, IL-6, and TNF) and cognitive function in healthy elderly humans in the general population (Baune et al., 2008b) were recently reported. More specifically, the results suggest that genetic variants of TNF may have protective effects in cognitive function such as perceptual speed (Baune et al., 2008b). In an animal model of cytokine-mediated cognitive function such as memory and learning, we were able to demonstrate that the presence of TNF under immunologically non-challenged conditions is essential for normal functions of memory and learning (Baune et al., 2008c). This research linking complex cognitive phenotypes and cytokine effects in the CNS is well based on previous evidence to suggest that cytokines play a role in normal CNS function at a cellular and molecular level (see Jankowsky & Patterson, 1999 for review). For instance, during health both IL-1 and TNF have been shown to act as neuromodulators, as well as, pro-inflammatory factors (Blatteis, 1990; Vitkovic et al., 2000a, 2000b).
Although these detrimental effects on cognitive function in both over-expressing and cytokine-deficient models suggest that cytokines play an important physiological role in the CNS at the molecular and cognitive level, it still remains to be fully understood how cytokines participate in the molecular and cellular mechanisms subserving complex CNS functions such as learning, memory, and cognition. Interestingly, growing evidence suggests that in particular the cytokines IL-1, IL-6, and TNF are involved in the molecular and cellular mechanisms subserving complex cognitive processes (Vitkovic et al., 2000a; Pickering & O’Connor, 2007; Viviani, Gardoni, & Marinovich, 2007). While direct and indirect evidence shows that IL-1, IL-6, and TNF may play a major role in synaptic plasticity, long-term potentiation (LTP), neurogenesis, and memory consolidation, the evidence for involvement with cognitive function is less conclusive for other cytokines such as interferon-gamma (IFNγ) (Baron et al., 2008), alpha(1)-antichymotrypsin (ACT) (McIlroy et al., 2000; Nilsson et al., 2004; Dik et al., 2005), and IL-2 (Beck, King, Huang, & Petitto, 2002; Beck et al., 2005a, 2005b).
Impaired neuroprotective and enhanced neurodegenerative effects of cytokines relevant to cognition and depression
During physiological conditions of early development and adulthood, cytokines have predominantly neuroprotective effects, with a wide array of functions subserving cognitive processes (Yirmiya & Goshen, 2011). The cytokine model of cognitive function, by McAfoose and Baune (2009), outlines the role of cytokines (e.g. TNF-α, IL-6, and IL-1β) in regulating neurobiological processes subserving cognition, i.e. synaptic plasticity, synaptic scaling, neurogenesis, neurotransmission, and long-term potentiation/depression (LTP/LTD). Under certain conditions, cytokine function can become neurodegenerative.
TNF-α is thought to exert its neuroprotective and neuroregenerative effects primarily via TNFR2 (p75; primarily neuroprotective and neuroregenerative pathway) and related signaling pathways (e.g. IkB kinase (IKK)/(NF-kB, transforming growth factor β-activated Kinase 1 (TAK-1), phosphoinositide 3-kinase (PI3K)-protein kinase B(PKB) and JNK), as opposed to the TNFR1-related neurodegenerative pathway (p55; primarily neurodegenerative (Santello & Volterra, 2012)). Signaling through TNFR2 promotes anti-apoptotic responses (e.g. Bcl-2, cIAP, xIAP, Bcl-XL) via the PI3K-PKB pathway, attenuates neuronal stress from ROS production, promotes proliferation of glial progenitor cells, as well as neuronal survival from glutamate-induced excitotoxicity via persistent NF-κB activation, and enhances HC neurogenesis (Fischer et al., 2011; Baune et al., 2012c). A study on double transgenic mice demonstrated that knockout of TNFR1 in Alzheimer transgenic APP23 mice resulted in reduction in brain Aβ plaques (He et al., 2007a), hence suggesting a neurodegenerative effect of TNFR1. A post-mortem study by Cheng and colleagues (Cheng, Yang, He, Li, & Shen, 2010) found increased TNFR1 and decreased TNFR2 protein levels in the frontal cortex and HC of Alzheimer’s disease (AD) brains compared with non-demented brains. This same study found in situ hybridization (ISH) data showed little change in mRNA levels of either TNFR in neurons of AD subjects; a functional binding study using 125I-TNF-α found R1 affinity was increased, whereas binding affinity to R2 was decreased.
IL-6 has been found to exert both neuroprotective and neurodegenerative effects via gp130 signaling and related pathways (e.g. janus kinase (JAK)/signal transducer and activator of transcription (STAT), MAPK/cAMP response element-binding (cAMP), Response Element-Binding (CREB), Ras-MAPK, PI3K (Baune et al., 2012b)). These mechanisms variously affect neural plasticity by their ability to induce processes of neurogenesis, such as gliogenesis, neuronal differentiation, cAMP, neural progenitor proliferation, and neuronal survival (Baune et al., 2012b). From a neuroprotective perspective, the binding of IL-6 to the IL-6Rα and subsequent activation of the STAT-3 pathway results in the production of the antioxidant, manganese-superoxide dismutase (Jung, Kim, & Chan, 2011). An approach using in vitro adenoviral gene delivery – to over-express IL-6 in cultured cerebellar granule cells – shows that IL-6 over-expression impairs granule cell adhesion and migration but has no effect on dendritic spines or granulate cell apoptosis (Wei et al., 2011). In rodent models, the difference between the pro- and anti-neurogenic effects of IL-6 may reflect differences in amounts and conditions used experimentally (Eyre & Baune, 2012).
A meta-analysis of 40 studies comparing peripheral blood cytokine concentrations between AD and healthy control subjects found higher levels of IL-6, TNF-α, IL-1β, transforming growth factor beta (TGF-β), IL-12, and IL-18 in AD subjects (Swardfager et al., 2010). There was no difference for IL-2, IL-4, IL8, or IFNγ. Another recent meta-analysis of 14 studies – measuring cerebrospinal fluid (CSF) cytokine levels in AD and healthy control subjects – found significantly higher levels of TGF-β in AD subjects compared with control subjects. There was no difference between groups for IL-6, TNF-α, and IL-1β. A number of prospective studies have found higher levels of PICs (i.e. IL-6, CRP, and TNF-α) are associated with increased risk of cognitive decline in aging populations (Koyama et al., 2013).
Impaired neuroprotective and enhanced neurodegenerative effects of microglia
Microglia contribute to the maintenance of the physiological state of the healthy brain through a number of neuroprotective mechanisms relevant to cognitive function (Sierra, Abiega, Shahraz, & Neumann, 2013). These mechanisms include phagocytosis of cellular debris and noxious stimuli within the CNS (e.g. Aβ protein and tau), breakdown of Aβ protein, production of neurotrophins and AICs, diversion of noxious stimuli away from neurons (e.g. excitotoxic glutamate), and modulation of synaptic plasticity (Streit & Xue, 2012; London, Cohen, & Schwartz, 2013). Streit and Xue (2012) suggest a microglial dysfunction hypothesis of AD, postulating impairment of neuroprotective function of these cells – via senescent deterioration – accelerates AD-related pathology, as also suggested by other authors (Liu et al., 2013; Morris, Clark, Zinn, & Vissel, 2013). Microglial dysfunction can occur due to aging, cellular senescence, and phenotypic changes and results in a failure of the above-mentioned neuroprotective processes, as well as the production of noxious proteins (i.e. PICs, ROS, and respiratory burst (Streit & Xue, 2012)). Dysfunction is thought to be represented by dystrophic morphology, i.e. shortened, gnarled, beaded, or fragmented cytoplasmic processes (cytorrhexis), as well as a loss of fine ramifications and formation of spheroidal swellings (Streit, Braak, Xue, & Bechmann, 2009). Recent data from aging bigenic APP/PS1 mice show that microglia can become dysfunctional via a significant reduction in expression of their Aβ-binding receptors and Aβ-degrading enzymes; however, they maintain their ability to produce PICs (Hickman, Allison, & El Khoury, 2008). In contrast, two recent rodent studies suggest that microglia do not influence Aβ deposition, nor facilitate its clearance (Grathwohl et al., 2009; Mildner et al., 2011). Selective depletion of microglia with the use of a lineage-ablation approach revealed no change in plaque formation, total Aβ load, or neurotoxicity in an AD mouse model (Grathwohl et al., 2009). These findings, however, may have been observed at a later stage of AD-related pathology when CNS cells were senescent.
Emotion regulation in the amygdala relevant to depression
The amygdala is part of the telencephalon (Murray, 2007) and appears as an almond-shaped neuronal core. It lies deep within the anterior temporal lobe (Murray, 2007) and is part of the limbic system. The limbic system is a principal region involved in social behaviors, such as affiliation, affection, and emotional intelligence (Killgore & Yurgelun-Todd, 2004). The amygdala is, when it comes to emotion regulation, a key region of the limbic system. Yet, it receives signals from most cortical fields (Stefanacci & Amaral, 2000; Ghashghaei & Barbas, 2002) as well as brain sections outside the limbic system, predominantly ventral forebrain structures (Phillips, Drevets, Rauch, & Lane, 2003a, 2003b), and usually returns them.
The amygdala consists anatomically of two different parts: the basolateral nucleus (BLA) and the central nucleus (CeA). While the BLA is characteristically considered as the sensory interface of the amygdala, the CeA is seen as the output region to other brain areas (Pare, Quirk, & Ledoux, 2004). The BLA can physiologically be subdivided in two groups of cells: one is involved in memory storage and the other in initial learning (Radwanska, Nikolaev, Knapska, & Kaczmarek, 2002). The BLA encodes the visionary relationship between impulses and primary reinforcers, such as food and fluids. In contrast to what was thought in the past, positive influences are as often encoded as negative ones (sad versus happy faces) (Murray, 2007).
Although it has been known for quite some time that the amygdala is involved in emotion processing (Weiskrantz, 1956), most recently accomplished research is based on its role in fear conditioning, which is a model of emotional learning in which psychological responses are being associated with an aversive stimulus (Bailey, Kim, Sun, Thompson, & Helmstetter, 1999; Davis & Whalen, 2001; Lin et al., 2001). However, the role of the amygdala in emotion processing and implicit learning is not limited to fear (Phelps & Ledoux, 2005) and includes other emotional conditioning as well. The amygdala is involved in the modulation and habituation of spatial memories and it even may favor stimuli that are predisposed leading to an emotional reaction (Phelps & Ledoux, 2005). However, it does not appear to be required in the modulation of memories of fear conditioning (Lee, Choi, Brown, & Kim, 2001). There are multiple other brain systems, including the limbic system, involved in the encoding and expression of this form of emotional learning (Phelps & Ledoux, 2005).
A large range of emotions are recognized to be related to processes in the amygdala, such as anger, happiness, sadness, pride, fear, anxiety, relief, and shame (Ramos & Mormede, 1998; Phelps & Ledoux, 2005), all relevant to depression. More complicated affective processes, including emotional learning, emotional memory, reward, rage moderation, emotional influences on attention and perception, sexuality, and social behavior, have also been linked to the amygdala (Mcmillian et al., 1995; Phelps & Ledoux, 2005; Murray, 2007). Previous research suggests that the amygdala not only mediates unconscious biases and preferences, but influences feelings such as hopes, dreams, ideas, beliefs, and dreads as well (Murray, 2007).
Effects of cytokines on the neurobiology and function of the amygdala
Cytokines act on the neurobiology of the amygdala through the onset of several mechanisms: the induction of other mediators such as chemokines, adhesion molecules, extracellular proteins, growth factors and other cytokines, and through the induction of cell death (John, Lee, & Brosnan, 2003). Churchill and colleagues (Churchill et al., 2006) have found that locally induced changes in the cytokine levels in the amygdala can be a result of the effect of peripheral cytokines. In this research it was clarified that peripheral cytokines induce brain cytokine mRNA, which is evident for the transcription and translation of cytokines in the amygdala. Also, Churchill and colleagues defined a relationship between peripheral cytokines and the cytokines which will ultimately be produced in the amygdala: IL-1β increases brain IL-1β mRNA, TNF-α increases brain TNF-α mRNA.
Certain cytokines will, after interaction with their receptor, induce a pathway of signal transduction within the amygdala. For TNF-α and IL-1β, it has been proven that both the NF-κB pathway and the mitogen-activated protein kinase (MAPK) pathway become activated (Saha, Liu, & Pahan, 2006). It is very likely that these pathways will also be activated by other cytokines (John et al., 2003). A recent study in MDD applying a combined pharmacogenetic and imaging approach suggests possible pathways linking inflammatory genes, regulation of emotions, and pharmacoresponse in MDD (Baune et al., 2010a). One of those pathways might be the anterior cingulate cortex (ACC), which plays a pivotal role in the experience and regulation of emotions (Phillips et al., 2003a) and has been strongly implicated in the pathophysiology of major depression (Phillips et al., 2003b). Several studies have demonstrated that pre- and subgenual anterior cingulate cortex (ACC) activity at rest (Mayberg et al., 1997; Pizzagalli et al., 2001), as well as reactivity to emotional faces (Chen et al., 2008) and pictures (Pizzagalli et al., 2001) predicts response to antidepressant medication in depression (also see for review Derubeis, Siegle, & Hollon, 2008). Along the same line, two recent studies demonstrated that responsiveness of the amygdala to emotional pictures (Canli et al., 2005) and words (Siegle, Carter, & Thase, 2006) is associated with a beneficial treatment outcome.
New data suggest a consistent finding in that the same genetic variants of IL-1b associated with non-remission after antidepressant treatment are also associated with decreased amygdala and ACC functions, which seem to represent neurobiological markers of a more severe course of disease and worse antidepressant treatment outcome (Baune et al., 2010a). Interestingly, this finding parallels the results of other imaging genetics studies describing a relative uncoupling of amygdala and ACC in carriers of potential risk alleles for unfavorable treatment response in depression (5-HTTLPR S allele (Pezawas et al., 2005) and MAO-A H alleles (Dannlowski et al., 2009)). Furthermore, these findings are in line with a study by Harrison and colleagues showing that a reduced amygdala–subgenual ACC coupling is associated with inflammation-induced mood decrease (Harrison et al., 2009). These findings on an impaired neurobiology of amydgala function associated with inflammation may exert important functional consequences on altered brain circuitries in depression. Depressed patients generally activate cognitive control circuitry the same as, or more than, controls during explicit down-regulation of negative emotion, but either did not show amygdala decreases, did not show the expected reciprocal amygdala–prefrontal relationship during regulation, or were unable to sustain those decreases (Etkin, Gyurak, & O’Hara, 2013). Hence, it can be proposed that inflammatory effects on amygdala function may exert far-reaching consequences on the function of larger brain circuitries affecting broadly emotion regulation and cognitive function in depression.
Conclusions
Recent developments in understanding the pathophysiology of clinical depression indicate a major role of inflammation in some groups of patients with depression. Inflammatory processes affect a variety of symptom domains of depression including cognitive processing, emotion regulation, sleep, appetite, and metabolic function in depression. Since inflammation and more broadly immune dysregulation in mood disorders involves a plethora of immune cells affecting brain function during different stages of depression, a phase-specific immune model of depression has recently been proposed (Figure 12.3). Such a model enables the understanding of a complex neurobiological and clinical relationship in depression in that immune processes affect symptom domains of depression in a phase-specific manner. It could assist to explain that immune dysregulation that is still present during remission may be a contributing factor to cognitive dysfunction frequently observed in patients with remitted depression. Hence, a phase-specific description of immunological dysregulation of depression may present an important conceptual advance in the complex relationship between immune dysfunction, depression, and cognitive dysfunction and their individual and combined effects on general and psychosocial function as described here. Moreover, immunomodulatory interventions might specifically be beneficial to MDD patients that present primarily with cognitive disturbance in addition to the common emotion processing impairments in conjunction with evidence of immune disturbance and/or increased inflammation. Novel anti-inflammatory and neuroprotective treatments may represent a new avenue in the treatment of patients with MDD and clinically relevant cognitive dysfunction in conjunction with immunological alterations. It can be assumed that treatment of depression and cognitive dysfunction targeting underlying impaired neurobiology such as inflammation would also enhance patient function in various domains such as social and interpersonal life, at work, and related performance and daily function.
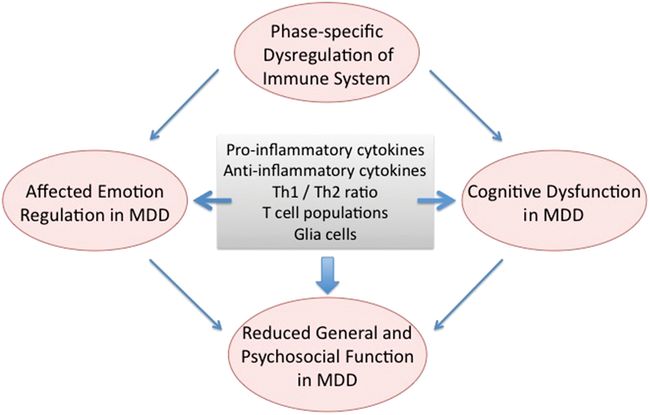
Immune dysfunction impacts on emotion deregulation and cognitive dysfunction in MDD.
References
Related posts:
 HPA axis and cognitive dysfunction in mood disorders
Are cognitive deficits in major depressive disorder progressive?
HPA axis and cognitive dysfunction in mood disorders
Are cognitive deficits in major depressive disorder progressive?
 Hot and cold cognition in major depressive disorder
Neuroscience of functional outcomes and treatment targets in major depressive disorder
Treatment of cognitive dysfunction in adults with major depressive disorder
Hot and cold cognition in major depressive disorder
Neuroscience of functional outcomes and treatment targets in major depressive disorder
Treatment of cognitive dysfunction in adults with major depressive disorder
 Measuring the mind: detecting cognitive deficits and measuring cognitive change in patients with depression
Measuring the mind: detecting cognitive deficits and measuring cognitive change in patients with depression
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


