CHAPTER 89 INFLAMMATORY MYOPATHIES
The inflammatory myopathies are a heterogeneous group of acquired diseases of skeletal muscle. They have in common the presence of varying degrees of muscle weakness and inflammation. Based on clinical, histological, and immunopathological criteria, they form three major groups: polymyositis, dermatomyositis, and sporadic inclusion-body myositis.1,2 Dermatomyositis is easily recognized by specific skin changes that occur early in the course of the disease. Sporadic inclusion-body myositis is easily suspected based on its slow progression, unique distribution of weakness and atrophy, characteristic muscle biopsy findings, and resistance to conventional immunotherapies.2,3 Polymyositis, however, remains a diagnostic challenge and is often misdiagnosed as inclusion-body myositis, dystrophy, or toxic or metabolic myopathy.4 The old assumption that dermatomyositis is like polymyositis without a rash, and inclusion-body myositis is like polymyositis without vacuoles, is overly simplistic and incorrect.5,6
CLINICAL PRESENTATION
Dermatomyositis
Dermatomyositis is seen in both children and adults, and more often in women than in men1 (Table 89-1). Juvenile dermatomyositis is the most common form of inflammatory myopathy in children.7,8 Although the most obvious manifestations are due to involvement of skeletal muscle and skin, rarely other organ systems are affected, including the gastrointestinal tract, heart, and lungs. The muscle weakness in dermatomyositis is classically proximal, symmetrical, and frequently progressive. It can vary from mild to severe, occasionally resulting in quadriparesis. The weakness usually develops slowly, over weeks to months, but in rare cases there is an acute onset. Patients usually have problems with physical tasks such as rising up from a chair or climbing steps, stepping onto a curb, lifting objects, or combing their hair. Fine motor movements that depend on the strength of distal muscles, such as manipulating small objects, are spared until late in the course of the disease. Involvement of the neck extensor muscles may lead to head drop. In advanced stages of the disease or during an acute course, patients might have respiratory muscle weakness or dysphagia, causing choking episodes. Facial muscles are spared and extraocular muscles are never affected.1 Sensation remains normal, and tendon reflexes are preserved but may be absent in severely weakened or atrophied muscles. Myalgia is not a common feature and occurs in less than 30% of the patients.1
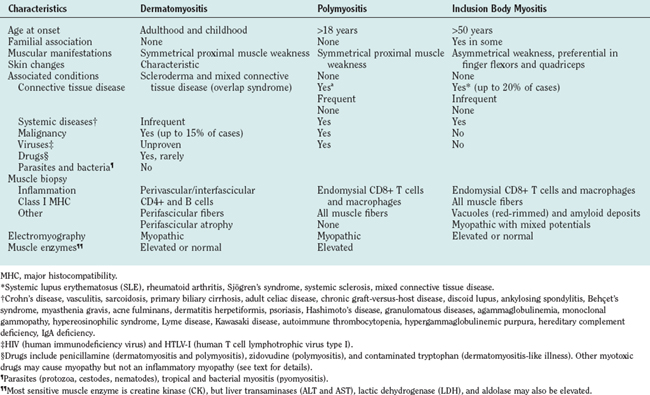
The cutaneous manifestations of dermatomyositis usually precede or accompany the weakness and include the following: erythematous (and later dry and scaly) lesions over the metacarpophalangeal, proximal interphalangeal, or distal interphalangeal joints (Gottron papules); a violaceous hue over the eyelids (heliotrope rash) and periorbital edema; periungual telangiectasia characterized by dilated capillary loops at the base of the fingernails, with irregular, thickened, and distorted cuticle; malar erythema; and erythematous scaly rash over the neck and upper back (shawl sign), anterior chest (V sign), and extensor surfaces of the extremities (Fig. 89-1). The rash can be exacerbated after exposure to the sun and is pruritic in some cases.1 At times, muscle strength appears normal—hence the term dermatomyositis sine myositis, or amyopathic dermatomyositis. When a muscle biopsy is performed in such cases, however, perivascular and perimysial inflammation can be seen. Juvenile dermatomyositis resembles dermatomyositis in adults, except for the presence of more frequent extramuscular manifestations. A child with evolving dermatomyositis is irritable, does not socialize, is uncomfortable, complains of fatigue, and has a red flush on the face with varying degrees of muscle weakness.9 A tiptoe gait due to flexion contracture of the ankles is also common in juvenile dermatomyositis.1 In 3% to 5% of children with juvenile dermatomyositis, the cutaneous manifestations of the disease are present in the absence of clinically evident muscle weakness: these patients are classified as amyopathic.10 Subcutaneous calcinosis is not an uncommon finding in juvenile dermatomyositis, especially when initiation of therapy is delayed, or when applied therapies have not been fully effective, resulting in chronicity of the disease, muscular atrophy, and joint contractures8,11,12 (Fig. 89-1).
Polymyositis
Polymyositis is usually seen after the second decade of life, rarely in children.2,1 It is manifested by muscle weakness of subacute onset, similar to what is seen in dermatomyositis but without any of the cutaneous manifestations. Polymyositis can mimic many other myopathies and remains a diagnosis of exclusion. The most common myopathy misdiagnosed as polymyositis is inclusion-body myositis. This disease is often suspected in retrospect, when a patient with presumed polymyositis has not responded to therapy.2 Other myopathies erroneously diagnosed and treated as polymyositis include acute necrotizing myopathies, toxic and endocrine myopathies, dermatomyositis sine dermatitis, certain dystrophies, and some slowly progressive myopathies starting in late childhood.1 One of the main reasons for misdiagnosing these disorders as polymyositis is that several myopathies, especially certain dystrophies such as Duchenne’s, Becker’s, fascioscapulohumeral, or dysferlinopathies, may also show prominent inflammation in their muscle biopsies.4 Thus, we have to apply new diagnostic criteria that more specifically characterize the type of inflammation seen in polymyositis, as discussed later.
Inclusion Body Myositis
Inclusion-body myositis is the most common form of inflammatory myopathy, especially in patients above the age of 50.2 The weakness and atrophy are usually observed first in the quadriceps femoris muscles of the legs, leading to instability of the knees and increased falls, and in the flexor digitorum profundus muscles in the forearm, resulting in difficulties with fine motor movements such as holding golf clubs, turning keys, or tying knots.13 The weakness and atrophy frequently affect the iliopsoas, triceps, biceps, and foot extensor muscles9 and it may be asymmetrical, resembling lower motor neuron disease or motor neuropathies. The facial muscles are very often affected even early in the disease. Dysphagia is common, occurring in up to 60% of inclusion-body myositis patients, and may lead to episodes of choking. Sensory examination is generally normal, although some patients have mildly diminished vibratory sensation at the ankles that is presumably age related.9 Disease progression is slow but steady, and most patients require assistive devices such as a cane, walker, or a wheelchair after several years.
Associated Clinical Manifestations
Inflammatory myopathies, in particular dermatomyositis and polymyositis, can have non–skeletal muscle manifestations. Constitutional symptoms such as fever, malaise, weight loss, arthralgia, or Raynaud’s phenomenon can occur in the subacute stages of dermatomyositis and polymyositis, especially when the disease is associated with another connective-tissue disorder.14 Cardiovascular abnormalities, including conduction defects, tachyarrhythmias, or myocarditis, are rarely seen during the active phase of the disease. Hypertension and heart failure are more frequently seen late in the course of the disease, probably as a result of longterm steroid therapy or lung disease.1 Pulmonary involvement may occur, either due to thoracic muscle weakness or interstitial lung disease. The latter is most commonly seen in patients with autoantibodies against t-RNA synthetases (Jo-1), signal recognition particle (SRP),15 or a mucin-like glycoprotein (KL-6).16,17 Antisynthetase syndrome refers to the presence of inflammatory myopathy, anti-Jo-1 antibodies, and interstitial lung disease, often in association with low-grade fever, subluxation of the phalangeal joints, Raynaud’s phenomenon, and thick cracked skin on the fingers (mechanic’s hands).18
Malignancy
Among the inflammatory myopathies, dermatomyositis is associated with increased incidence of malignant diseases, in up to 15% of the adult cases.19 The most common cancers are those of the ovaries, gastrointestinal tract, lung, breast, and non-Hodgkin lymphomas20 or nasopharyngeal cancer in Asian populations. Increased vigilance, with careful periodic clinical examination and the prudent use of appropriate laboratory studies, are required for early detection of cancer, especially in older patients, and during the first 3 years after disease onset.2,21
DIAGNOSIS
The criteria of Bohan and Peter22 used for several years, have now become obsolete because they do not distinguish polymyositis from inclusion-body myositis or certain muscular dystrophies (Table 89-2). The need for new criteria was recognized in 1991.2 The inclusion of simple immunopathology in processing the muscle biopsy specimens was recently emphasized as the best means of differentiating inflammatory from noninflammatory myopathies.1 The diagnosis is based on the characteristic clinical features of polymyositis, dermatomyositis, or inclusion-body myositis outlined earlier, combined with the triad of serum muscle enzyme levels, electromyography, and muscle biopsy. Imaging studies have a limited role in the diagnosis of inflammatory myopathies. Subcutaneous calcinosis, an uncommon finding in adult but more common in juvenile dermatomyositis, can be detected on plain radiographs.11 Magnetic resonance imaging (MRI) may reveal the involved muscles in inclusion-body myositis23,24 but is not needed for diagnostic purposes. The only usefulness of MRI is to help us select the appropriate site for biopsy, when the clinical pattern of weakness is not uniform or typical. We do not recommend routine MRI for our patients; we select the biopsy site on the basis of clinical assessment. In the chronic stages of these diseases, when muscle atrophy and fatty infiltration dominate the picture, MRI is of no diagnostic value.
Muscle Enzymes
Creatine kinase, aspartate and alanine aminotransferases, lactate dehydrogenase, and aldolase levels are usually increased in inflammatory myopathies, particularly in the active phases of the disease, and parallel the disease activity.1 Elevation of serum creatine kinase, although not specific to inflammatory myopathies, is a useful laboratory marker.9 Creatine kinase, however, may remain within the normal range in some patients, especially with dermatomyositis or inclusion-body myositis, and should not be used as the sole marker for determining disease activity.
Electrodiagnostic Studies
Needle electromyography shows myopathic potentials, characterized by increased spontaneous activity, fibrillations, complex repetitive discharges, and positive sharp waves. The voluntary motor units have low amplitude and short duration and are polyphasic. Mixed potentials (polyphasic units of short and long duration) indicating a chronic process or muscle fiber regeneration are often present in inclusion-body myositis.9 These findings, however, are not disease specific, as they can occur in any other active myopathy.9 At times, the presence of active myopathic potentials can distinguish exacerbation of the primary disease from steroid-induced myopathy.1
Muscle Biopsy
Muscle biopsy is the most important test for establishing the diagnosis, but it can also lead to misdiagnosis due to erroneous interpretation.1
Occasionally, due to the spotty nature of the inflammation, a repeat muscle biopsy may become necessary. This needs to be especially considered in the patients who meet the clinical criteria described earlier but whose initial muscle biopsies are nondiagnostic.1 We also recommend a repeat muscle biopsy when a patient carries the diagnosis of polymyositis but remains unresponsive to therapies.
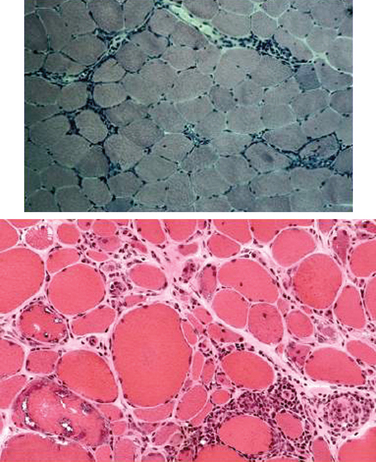
The histopathology of dermatomyositis is characterized by inflammation, which is predominantly perivascular and in the interfascicular septa rather than in the fascicles.3,9,25 Necrosis and phagocytosis of muscle fibers commonly occur in groups (microinfarcts) and involves the peripheral portion of the muscle fascicle. The resultant perifascicular atrophy (Fig. 89-2) is characterized by 2 to 10 layers of atrophic fibers and is diagnostic of dermatomyositis irrespective of the presence of inflammation. In the absence of typical features of inflammatory infiltrates or perifascicular atrophy, increased perifascicular class I major histocompatibility (MHC) expression, may be of diagnostic value.26 Biopsy of skin lesions in dermatomyositis shows perivascular inflammation in the dermis with CD4+ cells and dilatation of superficial capillaries in more chronic stages.1
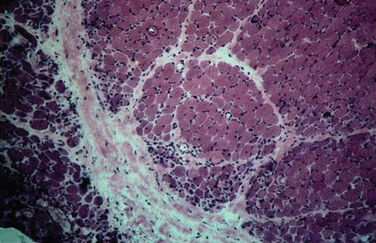
Figure 89-2 Perifascicular atrophy in a cross section of a muscle biopsy in a patient with dermatomyositis.
Muscle biopsies of patients with polymyositis show varying degrees of endomysial inflammation (Fig. 89-3). In polymyositis and inclusion-body myositis, the inflammation is characterized as primary to denote the partial invasion of still intact and nonnecrotic muscle fibers by activated CD8+ T lymphocytes. B lymphocytes form a minor fraction of perivascular infiltrates, but they are mostly absent from the endomysium.27,28 In chronic stages of the disease, there is an increase in connective tissue, which may react with alkaline phosphatase. Both invaded and noninvaded muscle fibers express class I MHC antigen on their surface29 (Fig. 89-4). The ubiquitous sarcolemmal expression of class I MHC antigen has now become essential in diagnosing polymyositis, not only because it is part of the specific lesion but also because it is not present in other non-immune myopathies and it is not affected by the use of immunosuppressive agents. The “CD8+/MHC-I complex” (MHC-I along with CD8+ cells) is specific for the inflammation in polymyositis and inclusion-body myositis and needs to be incorporated into the histological evaluation of polymyositis patients, in an effort to rule out the secondary inflammation seen in various dystrophies or toxic myopathies.1
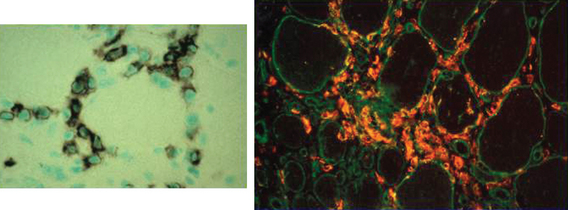
Muscle biopsies from patients with inclusion-body myositis show the same pattern of CD8/MHC-I expression as in polymyositis, even in the late stages of the disease. In inclusion-body myositis, however, in addition to inflammation, a number of muscle fibers not invaded by T cells contain single or multiple vacuoles, which seem to increase as the disease progresses4 (see Fig. 89-3). The vacuoles contain small, basophilic granules in their centers or against their walls and appear as “red-rimmed vacuoles” on the trichrome stain. Frozen sections and enzyme histochemistry are required for optimal visualization of vacuoles, because the granules dissolve on paraffin sections and might be easily overlooked if the biopsy is processed only with paraffin.4 Deposits of amyloid can be seen in vacuolated fibers as green birefringence on Congo red staining30 or as red dichroism when visualized with Texas red filters.31 Characteristically, the vacuolated fibers in inclusion-body myositis are not invaded by T cells, and those fibers that are partially invaded by inflammatory cells are never vacuolated.4 Vacuolated fibers, even the ones containing amyloid deposits, are not specific for inclusion-body myositis but can be seen in other chronic distal myopathies (e.g., myofibrillar, facioscapulohumeral, and dysferlin myopathies), and even in chronic neurogenic disorders such as old paralytic poliomyelitis.32 Other findings in inclusion-body myositis include ragged red fibers or cytochrome c oxidase–negative fibers, which contain abnormal mitochondria harboring multiple mitochondrial DNA deletions.33 These fibers are common in inclusion-body myositis and more frequent than expected for the patient’s age. On electron microscopy, the basophilic granules seen in cryostat sections represent membranous whorls of various sizes. In their vicinity, abnormal filaments 12 to 18 nm in diameter can be found. They have a tubular structure with a central lumen approximately 3.5 nm in diameter. On longitudinal view, these filaments often have a periodic cross-hatched appearance, which seems to be outside the axial core of the filament. The nature of the inclusion-body myositis filaments remains obscure. They immunoreact with various amyloid-related proteins, identical to those seen in Alzheimer’s disease, such as ubiquitin, phosphorylated tau, presenilin-1, apolipoprotein E (apoE), and others.34
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


