Intellectual Disability/Mental Retardation
Maris D. Rosenberg
Susan Vig
Definition and Terminology
2002 AAMR DEFINITION OF MENTAL RETARDATION
The following definition of mental retardation was endorsed in 2002 by the American Association on Mental Retardation: “Mental retardation is a disability characterized by significant limitations both in intellectual functioning and in adaptive behavior as expressed in conceptual, social, and practical adaptive skills. This disability originates before age 18.” (1). Implementation of the definition emphasizes the need to identify strengths, as well as limitations, of people with intellectual disability, and to develop profiles of supports that will optimize their functioning. The 2002 definition specifies that intellectual limitation is documented by an IQ approximately 2 standard deviations below the mean of a standardized intelligence test. Limitations in adaptive behavior (activities of daily living) are documented by the score on a test of adaptive behavior, based on information provided by a third party (parent, caregiver, or teacher).
CURRENT TERMINOLOGY
The term mental retardation is gradually being replaced by other terminology. In Great Britain, the terms “intellectual disability,” “learning disability,” and “learning difficulty” have long been preferred. Use of the term “learning disability” to represent intellectual and adaptive limitations has created confusion in the United States, where learning disabilities refer to academic deficits that are not consistent with intellectual potential.
In 2007, the American Association on Mental Retardation, a 130-year-old association representing disability professionals worldwide, changed its name to American Association on Intellectual and Developmental Disabilities. This change suggested that, in the United States and worldwide, the term “intellectual disability” might eventually replace the previous term “mental retardation.”
The term “developmental delay” is often used to characterize intellectual disability in young children. Most experts agree that it should be used to describe children younger than 3 years. Some experts have suggested that mental retardation should not be diagnosed in children younger than 5 years. In many educational settings, “developmental delay” is used for children up to 8 years of age.
In the following discussion, the term “intellectual disability” (ID) will be used instead of “mental retardation,” except where doing so would obscure meaning. Consistent with international usage, the term “children” includes adolescents.
Diagnostic Guidelines
In clinical settings, guidelines provided by the Diagnostic and Statistical Manual of Mental Disorders, Fourth Edition, Revised (2) are used to establish a formal diagnosis of ID (termed “mental retardation” in the diagnostic manual). This diagnosis is based on intellectual limitations (an IQ below 70 to 75) and deficits in adaptive behavior. Classification levels, based on IQ ranges, are mild (IQ of 50 to 55 to approximately 70), moderate (IQ 35 to 40 to 50 to 55), severe (IQ 20 to 25 to 35 to 40), and profound (IQ less than 20 to 25). The guidelines apply to children as well as adults, but do not specify the age at which this diagnosis may be made.
Prevalence
Prevalence estimates depend on how ID is defined. The prevalence of ID is generally thought to be approximately 2 ½% of the population (3). Increasing the IQ cutoff to 75 increases prevalence and means that more people will be eligible for publicly funded entitlements and services. According to Sattler (4), 85% of people with ID have mild ID, 10% have moderate, 3.5% have severe, and 1.5% have profound ID. More males than females have ID. Durkin and Stein reported male-to-female ratios of 1.1 to 1.8:1 for mild ID and 1.1 to 1.4:1 for severe ID (5). Gillberg and Soderstrom (6) reported a male-to-female ratio of 1.3:1, based on population studies, hypothesizing that this gender difference might be attributable to X-linked genetic mechanisms.
Within the United States educational system, reluctance to use the label “mental retardation” has affected prevalence estimates. As Baroff (7) pointed out, school systems may avoid using the term mental retardation for children with mild ID and associated academic deficits. Baroff reported that between 1977 and 1997, a 38% decline occurred in students classified as “mentally retarded” and an increase of 202% of students classified as “learning disabled” (7).
Etiology
Determination of the cause of ID has important implications for the patient and family. For the patient, an accurate diagnosis can provide the care provider with information about medical problems that may occur in the patient’s future, thus ultimately improving prognosis. For the family, the importance of identifying the cause of ID is threefold. First, parents often believe that their child’s problems were directly caused by things the parent did or did not do during or before the pregnancy, and establishing a biologic etiology can allay that guilt. Next, establishing a diagnosis can provide the family with a greater sense of control, when armed with the knowledge of cause, treatment options, and prognosis. Finally, establishing a diagnosis can provide the parents, siblings and extended family with a realistic assessment of risk of recurrence of the condition in future progeny. For all of these reasons, vigorous attempts should be made to identify the cause of ID in the individual patient.
In many patients, no specific etiology will ever be identified. According to Srour et al. (8), the diagnostic yield in patients with global developmental delay is approximately 40%. Factors associated with the ability to identify an underlying cause included female gender, abnormal pre/perinatal history, absence of autistic features, presence of microcephaly, abnormal neurologic examination, and dysmorphic features. No association was found between the severity of the delay and the identification of underlying etiology. Van Karnebeek et al. (9) and Battaglia and Carey (10), however, reported that an etiologic diagnosis
could be made in more than 50% of children with varying levels of ID based on history and examination alone, or history and physical findings with confirmatory laboratory studies. Serial evaluations over a period of several years have been suggested, in that clinical and behavior phenotypes can modify over time (11), and as new diagnostic techniques become available (12).
could be made in more than 50% of children with varying levels of ID based on history and examination alone, or history and physical findings with confirmatory laboratory studies. Serial evaluations over a period of several years have been suggested, in that clinical and behavior phenotypes can modify over time (11), and as new diagnostic techniques become available (12).
Recent advances in the area of genetics and genomics have made the determination of etiology increasingly possible. New technology has allowed the diagnosis of chromosomal anomalies associated with cognitive disabilities to be made with greater precision; molecular genetic techniques have broadened the scope of our understanding of mechanisms such as in mitochondrial inheritance, imprinting, uniparental disomy, and trinucleotide repeat disorders. Genomics, the study of the functions and interactions of all genes in the genome, has led to the understanding that complex disorders such as mental retardation are due to the interaction of genes, epigenetic (those that modify the expression of a gene) factors, and environmental factors. All this has rendered the genetic evaluation central to the assessment of an individual with cognitive delay. The reader is referred to several excellent reviews of current technology (13, 14, 15).
The Clinical Assessment
Despite the rapid expansion of knowledge in genomic medicine, the medical assessment of the child initially seen with ID, geared toward determining an etiology, is currently initiated in the clinical setting, not in the laboratory. Three recent statements by the American College of Medical Genetics (11), American Academy of Neurology and Child Neurology Society (16), and American Academy of Pediatrics Committee on Genetics (12) discussed the approach to the evaluation of children with ID and share many common elements. Each emphasizes the need for a careful history beginning with pregnancy, including exposure to teratogens, and perinatal course. Family history should include a pedigree of three generations or more, detailing the presence of developmental disorders (with careful attention to distribution e.g., suggestion of X-linked inheritance), congenital malformations, psychiatric illness, and frequent miscarriages/stillbirths/early childhood deaths. Physical and neurologic examinations focus on growth parameters, dysmorphic features (documenting minor anomalies with detailed measurements, skin changes with Wood’s lamp), and neuromuscular and behavioral features that suggest a specific syndrome.
Genetic Testing
Chromosomal anomalies have been detected in individuals with ID with a frequency ranging between 9% and 36% by using high resolution (>650 bands) karyotyping (12). Van Karnebeek et al. (9) reported such abnormalities to be present in all levels of ID and in both genders, with a median frequency of 1 in 10, with a higher number of anomalies (more than six) significantly increasing the likelihood of a chromosomal anomaly (9). Shevell et al. (16) reported the range of chromosomal anomalies to be between 2.3% and 11.6% and suggested “… cytogenetic testing is indicated in the evaluation of the child with developmental delay even in the absence of clinical features or dysmorphic features suggestive of a syndrome.”
Fluorescence in situ hybridization (FISH) techniques, by using fluorescently labeled cDNA probes, have been used to increase the yield of genetic testing for the cause of ID. FISH can be used to confirm a clinical suspicion of a microdeletion syndrome, many of which have specific behavioral phenotypes, such as Williams (del 7p), Prader-Willi/Angelman
syndrome (del 15p), catch 22 syndrome (del 22q), and Smith-Magenis syndrome (del 17p). In addition, rearrangements leading to deletions of the functional end of the chromosome (subtelomeric region) are thought to be responsible for a significant number of structural chromosomal abnormalities not previously detected with routine karyotype analysis. (17). Biesecker et al. (18) found a 6% rate of subtelomere abnormalities detected by FISH in a variety of subjects evaluated for ID, growth retardation, major and minor anomalies, and familial versus sporadic occurrence. DeVries et al. (19) proposed a five-item checklist designed to increase the yield of subtelomeric studies. The presence of three or more features (family history of ID, prenatal-onset growth retardation, postnatal poor growth/overgrowth, two or more facial dysmorphic features, or one or more nonfacial dysmorphic features and/or congenital abnormalities) would allow the exclusion of 20% of cases without missing a subtelomeric abnormality.
syndrome (del 15p), catch 22 syndrome (del 22q), and Smith-Magenis syndrome (del 17p). In addition, rearrangements leading to deletions of the functional end of the chromosome (subtelomeric region) are thought to be responsible for a significant number of structural chromosomal abnormalities not previously detected with routine karyotype analysis. (17). Biesecker et al. (18) found a 6% rate of subtelomere abnormalities detected by FISH in a variety of subjects evaluated for ID, growth retardation, major and minor anomalies, and familial versus sporadic occurrence. DeVries et al. (19) proposed a five-item checklist designed to increase the yield of subtelomeric studies. The presence of three or more features (family history of ID, prenatal-onset growth retardation, postnatal poor growth/overgrowth, two or more facial dysmorphic features, or one or more nonfacial dysmorphic features and/or congenital abnormalities) would allow the exclusion of 20% of cases without missing a subtelomeric abnormality.
Molecular genetic testing for fragile X syndrome has been recommended for children with undiagnosed ID, particularly in the presence of a family history or physical features suggestive of this diagnosis (11,12,16). Fragile X syndrome is the most common inherited form of ID occurring in 1 of 4,000 males and 1 of 8,000 females. The prevalence of the fragile X mutation is reported to vary with severity of retardation, ranging from 1% in borderline IQ or mild ID to 4.1% in more-severe degrees of MR (9). De Vries et al. (19) proposed a seven-item clinical checklist that increased the molecular diagnostic yield to 7.6%. Features include a family history of ID, long jaw or high forehead, large or protuberant ears or both, hyperextensible joints, soft and velvety palmar skin with redundancy on the dorsum of the hands, testicular enlargement, and behaviors of initial shyness and lack of eye contact followed by friendliness and verbosity. Molecular testing also is suggested to confirm diagnoses in syndromes in which the gene has been identified, such as in Rett syndrome, or in cases in which an atypical presentation of a known syndrome is suspected.
Not yet widely clinically available, the newest diagnostic technology, molecular karyotyping using microarray comparative genomic hybridization, allows the detection of abnormal copy numbers of DNA sequences throughout the entire human genome. This approach can reveal causative chromosomal microdeletions or duplications or both in 10% to 20% of patients with unexplained MR and congenital malformations (14). Nucleic acid probes are deposited on microarray platforms, which, when hybridized to test DNA, detect sequence variations known as single-nucleotide polymorphisms (SNPs). Microarrays have been developed to analyze hundreds of thousands of SNPs that compose an individual’s genome. The challenge lies in determining those copy-number alterations that cause disease versus those that are unique to the individual but have no clinical consequence. CGH will not replace routine chromosomal analysis, in that chromosomal rearrangements such as inversions or translocations, as well as low-level mosaicism may be missed by this technology (14).
Neuroimaging
CT and MRI studies are not considered mandatory in the evaluation of children with ID. Moeschler et al. (12), in their review of the literature, reported abnormal findings in approximately 30% of patients with ID. However, such findings led to an etiologic or syndrome diagnosis in only up to 3.9% of patients. Diagnostic yield was higher with an abnormal neurological examination. Shevell et al. (16) thus recommend neuroimaging (MRI preferable to CT) in cases with abnormal neurological examination (focal motor findings or microcephaly). Curry et al. (11) also do not recommend routine neuroimaging in the normocephalic patient without clinical neurological signs.
Metabolic Testing
Routine metabolic screening, in the absence of suggestive clinical history or abnormal laboratory findings, is reported to have a diagnostic yield of less than 1% (9,11,12,16). Targeted metabolic studies are suggested with specific clinical or laboratory findings such as growth failure, recurrent unexplained illness, neurologic findings, specific dysmorphic features, or laboratory abnormalities (lactic acidosis, hyperammonemia).
Figure 12.1 details the diagnostic algorithm published by the AAP Committee on Genetics based on the approach suggested by van Karnebeek et al. (9).
Characteristics of Children with ID
PRESENTATION
The presentation of intellectual disability in children varies by level of severity; in general, the more severe the level, the earlier the age at presentation, controlling for environmental factors. Failure to achieve expected motor milestones in the first year of life generally corresponds to levels of moderate retardation and below; a thorough medical workup for causal factors is indicated. Mild mental retardation is typically manifest by a delay in attainment of language milestones in toddlers and preschoolers. Such a presentation can also be consistent with a developmental language disorder, in which delays are specific to the language domain and not global in nature, as in ID. Multidisciplinary assessment is indicated for proper differentiation, and formal audiological assessment is essential to rule out a contributing hearing loss. Young school-age children, who begin to struggle with academic demands in kindergarten and early elementary school, may be demonstrating a slower rate of learning associated with borderline intellectual functioning. Once again, multidisciplinary assessment is indicated to differentiate such children from those who are functioning in the average range of intelligence, but whose learning many be compromised by specific deficits consistent with a learning disability.
INTELLECTUAL LIMITATIONS
The primary characteristic of ID is cognitive limitation, indicated by an IQ below 70 to 75 (2 standard deviations below the mean of an intelligence test). In thinking about children, it is useful to consider their mental ages. Those with mild ID function at about two thirds of their chronological ages. Those with moderate, severe, and profound ID function at approximately one half, one third, and below one fourth of their chronological ages, respectively. Within the developmental disabilities field, individuals with IQs below 50 are often said to have “significant” or “severe” impairment. (The term “severe” can thus be used in a technical sense to indicate a particular classification level, or less technically, to refer to significant impairment.) IQs between approximately 70 and 79 (and sometimes between 70 and 84, depending on whether test norms or standard-deviation norms are used for classification), indicate borderline intelligence. Although they are not formally diagnosed with ID, children with borderline intelligence experience many of the same challenges as those with mild ID, and are at high risk of developing academic and behavior problems (6).
Children with ID reach plateaus beyond which further intellectual development is not anticipated. Sattler (4) indicated the approximate mental ages to be expected at adulthood: 8.3 to 10.9 years for mild, 5.7 to 8.2 years for moderate, 3.2 to 5.6 years for severe, and less than 3.2 years for profound ID. (See references 4, 20, and 21 for specific skills expected at different ages and levels of ID.)
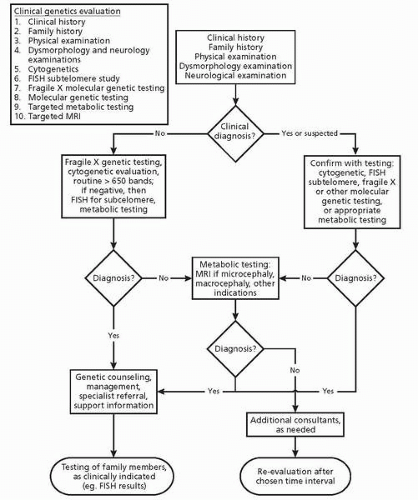 ▪ FIGURE 12.1. Diagnostic algorithm published by the AAP Committee. (Reprinted with permission from Moeschler JB, Shevell M, the Committee on Genetics. Clinical genetic evaluation of the child with mental retardation or developmental delays. Pediatrics 2006;117:2304-2316. |
LEARNING AND INTERACTION STYLES
In addition to having subaverage intelligence and mental ages below their chronological ages, children with ID exhibit learning and interaction styles that differ from those of peers with typical development. During the preschool years, children with ID are apt to show less
curiosity, less interest in discovering the function of objects, and more-restricted play than other children. They often experience difficulties establishing and developing friendships with other children, engage in higher levels of solitary play, and exhibit more negativity and discontent in peer interaction (22). Children of all ages with ID learn more slowly and forget more readily than other children. They may revert to earlier patterns and fail to use the new skills they have acquired. Children with ID do not generalize well and do not benefit from incidental learning opportunities. Teaching must be explicit and include frequent review. Children with ID may have trouble interpreting and prioritizing social information. Although eager for social approval, their judgment is apt to be poor, resulting in vulnerability to exploitation or maltreatment.
curiosity, less interest in discovering the function of objects, and more-restricted play than other children. They often experience difficulties establishing and developing friendships with other children, engage in higher levels of solitary play, and exhibit more negativity and discontent in peer interaction (22). Children of all ages with ID learn more slowly and forget more readily than other children. They may revert to earlier patterns and fail to use the new skills they have acquired. Children with ID do not generalize well and do not benefit from incidental learning opportunities. Teaching must be explicit and include frequent review. Children with ID may have trouble interpreting and prioritizing social information. Although eager for social approval, their judgment is apt to be poor, resulting in vulnerability to exploitation or maltreatment.
OTHER CHARACTERISTICS
Although some children with ID have dysmorphic features, many have an unstigmatized physical appearance. Most have adequate motor skills. It should be noted that, in assessment, failure of motor tasks may represent failure to comprehend cognitive aspects of the tasks. Many children with ID (particularly those with concurrent autism-spectrum symptoms) have isolated areas of stronger ability, often termed “savant” or “splinter” skills. (See reference 23 for a discussion of savant capacities.) Young children may be unusually good at labeling colors, shapes, letters, and numbers; some may read words. Older children may have well-developed calendar, mental-calculation, drawing, or musical talents. Although such achievements are impressive, they remain isolated and do not carry over into other aspects of cognitive ability.
Epilepsy in ID
The incidence of epilepsy in children with mental retardation is significantly higher than that in the general population (20% to 40% in children with ID and CP) (24, 25, 26); 8% to 18% in mild MR, and 30% to 36% in severe MR (27). The type of epileptic syndrome and the frequency and severity of seizures are related to the underlying cause of ID. The principles for management of epilepsy in individuals with ID are accurate diagnosis and classification of seizure type, consideration of associated medical conditions (e.g., tuberous sclerosis, cerebral palsy), use of appropriate medication and careful monitoring for side effects, and provision of information and support for the patient and family (27).
Seizures may exacerbate the functional deficits seen in individuals with ID. In the Isle of Wight study, up to 56% of children with MR and epilepsy exhibited comorbid psychiatric disorders as opposed to 30% in children with mental retardation alone (28). Matson et al. (29) stated that those individuals with ID and a diagnosis of seizure disorder were found to have significantly lower social and adaptive skills when compared with developmentally disabled controls with no seizures.
A number of epileptic syndromes are seen in association with ID. Lennox-Gastaut syndrome involves multiple types of intractable seizures and is associated with cognitive impairment (30). West syndrome involves the triad of infantile spasms, hypsarrhythmia on interictal EEG, and mental retardation, associated with a variety of etiologies and most commonly first seen in the first year of life (31).
Diagnosis of seizure disorder may be complicated in individuals with intellectual disability because of the presence of aberrant behavior, stereotypies, or involuntary movements that can be mistaken for seizures. Conversely, such movements/behaviors can be mistakenly assumed to be seizure related. In such cases, simultaneous EEG monitoring is indicated. Individuals with disabilities have more difficulty in achieving seizure control,
greater likelihood of clusters of seizures, prolonged seizures, and status epilepticus. Despite lessening the frequency or intensity of seizures, antiepileptic medications have the potential to cause cognitive slowing or behavior disturbances or both, which significantly affect the quality of life. The new generation of antiepileptic drugs, such as lamotrigine, topiramate, zonisamide, levetiracetam, and oxcarbazepine, promise better seizure control with fewer idiosyncratic reactions and side effects. Nonetheless, careful monitoring and attention to potential interactions with medications prescribed for other purposes is indicated. For detailed discussions of the use of these medications, the reader is referred to reviews by Shields (24), Huber and Seidel (25), Pellock and Morton (26), and Santosh and Baird (27).
greater likelihood of clusters of seizures, prolonged seizures, and status epilepticus. Despite lessening the frequency or intensity of seizures, antiepileptic medications have the potential to cause cognitive slowing or behavior disturbances or both, which significantly affect the quality of life. The new generation of antiepileptic drugs, such as lamotrigine, topiramate, zonisamide, levetiracetam, and oxcarbazepine, promise better seizure control with fewer idiosyncratic reactions and side effects. Nonetheless, careful monitoring and attention to potential interactions with medications prescribed for other purposes is indicated. For detailed discussions of the use of these medications, the reader is referred to reviews by Shields (24), Huber and Seidel (25), Pellock and Morton (26), and Santosh and Baird (27).
In instances in which medications do not achieve optimal control or in which side effects limit the effectiveness of pharmacotherapy, other treatment options such as ketogenic diet, vagus nerve stimulation (VNS), and epilepsy surgery can be considered. A ketogenic diet has been proven effective for medically refractory seizures (32,33). It has also been reported to improve sleep quality in children with therapy-resistant epilepsy (34). Vagus nerve stimulation has been reported effective in drug-resistant partial epilepsy (35) and in generalized and mixed forms (36). VNS avoids cognitive slowing, sedation, or behavioral effects. It is approved for patients older than 12 years and can be used as an adjunct to antiepileptic drug therapy. Effectiveness may be delayed for up to 1 to 2 years after implantation; effects on voice quality or swallowing are possible, and periodic readjustment of settings is required. A recent report suggests synergistic effects with the use of the ketogenic diet and VNS in children with medically refractory epilepsy (37).
A number of surgical procedures are considered for children with ID whose seizures are refractory to other treatments. Focal cortical resection can be successful in cases in which a defined epileptogenic zone can be safely removed without affecting the patient’s functional status. It is most successful when performed early, after two or three failed attempts at control with medication (24). Advances in imaging techniques, EEG, and cortical mapping have contributed to the success of these procedures. Other surgical procedures to be considered include temporal lobectomy, hemispherectomy, callostomy, and subpial transection.
Dual Diagnosis
PREVALENCE
“Dual diagnosis” refers to the co-occurrence of intellectual disabilities and psychiatric disorders or behavior problems. Prevalence estimates of comorbid psychopathology vary widely, because of different ways of defining and measuring psychopathology, types of behavior problems included or excluded, characteristics of samples studied, and whether study participants reside in institutions or in the community (38, 39, 40). The prevalence of clinically significant behavior problems or psychiatric disorders in people with ID (including children) has been reported as 30% to 70% (41); 23% to 38% (42); 37% (43,44); 50% (45); 40% to 60% of children (39); 40% (46); 25% (47); more than 40% (48); 38% of preschool children (49); 39% of children and adolescents (3), 30% to 50% (50); and 37% of children (51). Higher rates are reported for children with ID than those for other children (3). Although the presence of comorbid psychopathology is well documented for children with ID, mental health services are not necessarily provided for them. In a study by Dekker and Koot (43), only 27% of children with a dual diagnosis received mental health services.
Related posts:
 Developmental Characteristics of Sleep and Sleep Disorders in Children
Neuropsychiatric Aspects of Pediatric HIV Infection
Movement Disorders in Children and Adolescents
Neuroradiological Imaging in Children
Neurofibromatosis Type 1 and Tuberous Sclerosis Complex
The Diagnostic Approach to Neuropsychiatric Presentations
Developmental Characteristics of Sleep and Sleep Disorders in Children
Neuropsychiatric Aspects of Pediatric HIV Infection
Movement Disorders in Children and Adolescents
Neuroradiological Imaging in Children
Neurofibromatosis Type 1 and Tuberous Sclerosis Complex
The Diagnostic Approach to Neuropsychiatric Presentations
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


