, Jean Paul G. Vonsattel2, Helmut Heinsen3, 4 and Horst-Werner Korf5
(1)
Dr. Senckenbergisches Chronomedizinisches Institut, Goethe University Frankfurt, Frankfurt, Germany
(2)
Medical Center Neurological Institute, Columbia University, New York, NY, USA
(3)
Division Psychiatic Clinic Morphological Brain Research Unit, Julius Maximilians University Würzburg, Würzburg, Germany
(4)
University of Sao Paulo Medical School, Sao Paulo, Brazil
(5)
Dr. Senckenbergisches Chronomedizinisches Institut, Goethe University, Frankfurt, Frankfurt, Germany
8.1 Axonal Transport Mechanisms in Healthy Nerve Cells
Neurons are extremely polarized cells with a complex and unique morphology, as well as long processes (axon and dendrites) that extend far away from the nerve cell body. This architectonic feature of polarization with afferent (i.e., dendrites) and efferent processes (i.e., axons) requires efficient communication between the body of nerve cells and their periphery and makes nerve cells particularly dependent on functionally intact, sufficient, and timely axonal and/or dendritic intracellular transport processes over long distances. These intra–axonal trafficking processes guarantee bidirectional transport of subcellular compartments, including membrane organelles to or from the nerve cell processes, and enable the supply for specific action sites in the axon (e.g., nodes of Ranvier, axon terminal, synapses) and the disposal of vital cell organelles, membrane structures, synaptic vesicles, soluble neuronal proteins, or ion channels. Therefore, these targeted intraneuronal transport processes represent an essential prerequisite for the survival of the nerve cells of the human brain. The functional maintenance of nerve cells depends largely on the sufficient, locally correct, and precisely timed anterograde axonal transport of proteins and cell organelles from the cell body. In addition, the proper nerve cell function requires the retrograde trafficking of worn-out cell organelles back from the terminal regions to the cell body (Fig. 8.1) (De Vos et al. 2008; Gunawardena and Goldstein 2005; Li and Conforti 2013; Millecamps and Julien 2013). Long-range microtubule–based transport of cargos is the main mechanism (1) to deliver and target cellular components, organelles, and proteins from the cell body to their peripheral action site and/or (2) to remove them in the case of exhaustion. This long-range microtubule-based transport is accomplished by two major components: (1) “engines” or main microtubule-based molecular motors (i.e., kinesin, dynein) which need energy from ATP hydrolysis to move cargos along the microtubule cytoskeletal tracks and (2) neuronal microtubules, which represent the “rails” on which molecular motors loaded with cargos can run. Microtubules are very important in a number of cellular processes and are involved in maintaining the structure of the cell and, together with microfilaments and intermediate filaments as well as microtubules, form the cytoskeleton within the nerve cell’s cytoplasm. Neuronal microtubules are long, hollow cylinders made up of polymerized α– and β–tubulin dimers, are among the core structures of the neuronal cytoskeleton, and are crucial for the maintenance of its structure as well as for organizing motility and transport of intracellular constituents. Microtubule–associated proteins (MAP) like the cytoskeletal protein tau bind to and stabilize neuronal microtubules, promote their correct polymerization and assembly, and maintain their structural integrity. Neuronal microtubules are highly dynamic structures capable to undergo rapid periods of growth and shrinkage to generate force and enable the motor proteins kinesin and dynein to transport neuronal organelles and other cellular components, as well as neuronal proteins (Fig. 8.1) (De Vos et al. 2008; Gunawardena and Goldstein 2005; Li and Conforti 2013; Millecamps and Julien 2013).
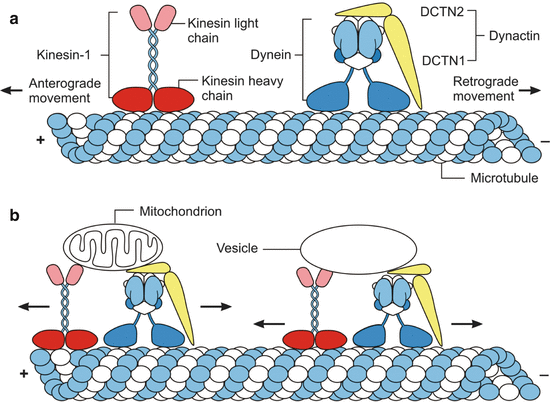
Fig. 8.1
The axonal transport mechanisms in healthy nerve cells. (a) The long-range microtubule-based axonal transport of cargos is the main mechanism to deliver and target cellular components, organelles, and proteins from the neuronal cell body to the action sites in the periphery and/or to remove them. This transport is accomplished by “engines” or main microtubule-based molecular motors (i.e., kinesin or dynein which generates force from ATP hydrolysis to move cargos along the microtubule cytoskeletal tracks) and neuronal microtubules, which represent the “rails” on which cargo-loaded molecular motors can run. The multimeric kinesin and dynein complexes have ATPase activity, can bind to the neuronal microtubule rails or tracks as well as to different cargos, and therefore represent the engines or molecular motors of the axonal transport that govern the microtubule-based axonal transport of most cargos and propel these cargos along neuronal microtubule cytoskeletal tracks. Kinesin–1 is composed of two heavy chains (red) and two light chains (light red) and moves cargos in the anterograde direction along axons (i.e., toward the axon tip and the plus end of microtubules, +). The heavy chains of kinesin-1 are able to bind to microtubule rails and contain its ATPase activity, while its light chains bind the cargos. Cytoplasmic dynein is a multi-subunit protein complex that moves cargos in the retrograde direction (i.e., toward the minus end of the microtubules, −) and is built up by heavy chains (blue) and intermediate, light intermediate, and light chains (light blue), as well as by dynactin subunits (such as DCTN1 and DCTN2; yellow) implicated in cargo binding. While the heavy dynein chains harbor ATPase activity and bind to neuronal microtubules, all other protein chains of dynein are involved in cargo and dynactin binding. (b) Cargos (e.g., mitochondria and vesicles) are attached to the molecular motor kinesin-1 by its light chains (light red) and to the engine dynein by its intermediate, light intermediate, and light (light blue) chains, as well as by associated dynactin subunits (yellow) (Modified according to Millecamps and Julien (2013), (Figure 1, page 163); with kind permission from Nature Publishing Group). Abbreviations: DCTN1 dynactin subunit 1, DCTN2 dynactin subunit 2
The molecular motors kinesin and dynein have ATPase activity, can bind to the neuronal microtubule rails as well as to different cargos, and are crucial role for the movement of cargos along cytoskeleton tracks. Kinesin–1 is the best known member of the kinesin superfamily, consists of two kinesin heavy and two kinesin light chains, and harbors a catalytic motor domain. This motor domain binds to microtubule rails, contains the ATPase activity, and together with a short neck linker region confers processivity and directionality. The tail of kinesin-1 together with its light chains binds the cargo and thus regulates the motor activity of kinesin-1. Cytoplasmic dynein, likewise, is a multi-subunit protein complex built up by two heavy chains, as well as intermediate, light intermediate, and light chains. While the heavy dynein chains harbor ATPase activity and, via the motor domains, can bind neuronal microtubules, all other dynein protein chains are implicated in cargo and dynactin binding (Fig. 8.1).
Most of the known molecular motors are involved in cargo trafficking in only one direction. While molecular motors of the kinesin family mediate axonal transport toward the synapse (i.e., anterograde axonal transport), molecular motors of the dynein family mediate transport of most cargos toward the cell body (i.e., retrograde axonal transport). Based on the bulk speeds of saltatory cargo movement, the axonal transport mechanisms in both anterograde and retrograde directions are classically divided into fast and slow components. While cargos such as vesicles, mitochondria, and endosomes move by fast axonal transport at speeds of ∼1 μm/s, neuronal cytoskeletal components move in slow axonal transport at speeds of ∼1 mm/day. Fast as well as slow components of anterograde and retrograde axonal transport processes are mediated by the same molecular motors kinesin and cytoplasmic dynein. Their different speeds result from different pauses between kinesin- and/or dynein-based cargo movements in the anterograde and retrograde directions (Fig. 8.1) (De Vos et al. 2008; Gunawardena and Goldstein 2005; Li and Conforti 2013; Millecamps and Julien 2013).
Damage to and disruption of axonal transport in human neurodegenerative diseases can occur via four mechanisms and contribute to dysfunctions and/or demise of affected nerve cells: (1) impairments of the engines or molecular motors, (2) damage to the microtubule rails, (3) damage to the cargos, and (4) loss of energy supply caused by mitochondrial damage (Fig. 8.2) (De Vos et al. 2008; Gunawardena and Goldstein 2005; Li and Conforti 2013; Millecamps and Julien 2013). Mutations in the main microtubule-based motors kinesin-1 and cytoplasmic dynein which inhibit their activity as molecular carriers, as well as abnormal phosphorylation of kinesin-1, are among the known molecular causes that may lead to impairments of the engines or molecular motors of axonal transport mechanisms in human neurodegenerative diseases. In human neurodegenerative diseases damage to microtubule rails can result from inhibition, abnormal bundling or destabilization of microtubules, damage to the axonal trafficking of microtubules themselves, and/or impaired interactions of microtubules with molecular motors. Each of these damages can lead to the deregulation of the dynamic properties of microtubule rails and to the disruption of cargo transport via axonal transport mechanisms. Mechanisms that lead to axonal transport defects in neurodegenerative diseases via damage to cargos and disruption of molecular motor–cargo interaction include abnormal phosphorylation or misassembly of cargos (e.g., neurofilaments). Finally, axonal transport deficits caused by mitochondrial damage are associated with a reduced anterograde transport of mitochondria and vesicles which results in a diminution of mitochondria in axons and decrease of ATP supply to molecular motors and, in turn, leads to decreased anterograde and retrograde movement of other axoplasmic cargos. The reduced ATP production caused by mitochondrial dysfunction might be part of a vicious circle that ultimately leads to dying-back of axons (Fig. 8.2) (De Vos et al. 2008; Gunawardena and Goldstein 2005; Li and Conforti 2013; Millecamps and Julien 2013).
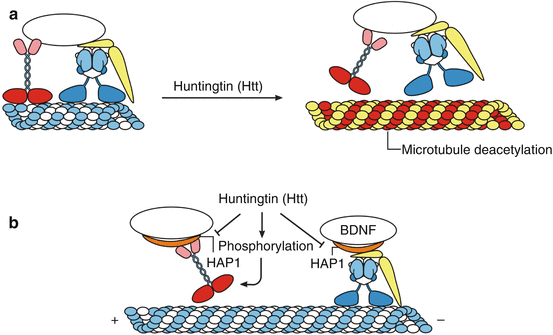
Fig. 8.2
Impairments of axonal transport mechanisms in Huntington’s disease (HD). In human neurodegenerative diseases damage to and disruption of axonal transport mechanisms can occur via four mechanisms which ultimately contribute to dysfunctions and/or demise of affected nerve cells: (1) impairments of the engines or molecular motors, (2) damage to the microtubule rails, (3) damage to the cargos, (4) injury of the energy supply caused by mitochondrial damage. Although the exact pathophysiological mechanisms are not fully understood, the following functional restrictions of mutant huntingtin (Htt) are currently considered to contribute to impairments of axonal transport processes in Huntington’s disease (HD): (1) reduced acetylation of the α-tubulin components of microtubules, which results in an impairment of kinesin-1 and dynein binding to and release of these molecular motors from the microtubule rails, (2) impaired binding and interaction of mutant Htt with the Htt-associated protein HAP1 that infers with axonal vesicular trafficking and organelle transport, (3) abnormal phosphorylation of kinesin-1 that reduces the binding of this molecular motor to and interaction with the microtubule tracks, and (4) abnormal anterograde and retrograde trafficking of mitochondria. (a) In HD, axonal transport defects may be caused by disruption of the binding of the motor proteins to microtubules or cargos. Mutant Htt promotes deacetylation of the α-tubulin components of microtubules and results in an impairment of kinesin-1 and dynein binding to and release of these molecular motors from the microtubule rails. (b) Microtubule track binding of kinesin-1 is also prevented by phosphorylation and interaction of mutant Htt with the Htt-associated protein HAP1 (orange). HAP1 is an adaptor protein that interacts with kinesin-1 light chains and the dynein-associated dynactin subunit 1 (DCTN1, yellow). Disruption of HAP1-mediated axonal transport also includes delivery of brain-derived neurotrophic factor (BDNF) (Modified according to Millecamps and Julien (2013), (Figure 2, page 16); with kind permission from Nature Publishing Group). Abbreviations: BDNF brain-derived neurotrophic factor, HAP1 huntingtin-associated protein1, Htt huntingtin
Mutiple evidences suggest that HD is among the polyglutamine diseases which go along with disruptions of axonal transport mechanisms and that damage to these transport processes is primarily associated with impaired functionality of mutant huntingtin (Htt). Although the exact pathophysiological mechanisms are not fully understood, the following functional restrictions of mutant Htt are currently considered to contribute to impairments of axonal transport processes in HD: (1) impaired binding and interaction of mutant Htt with the Htt-associated protein HAP1 that interferes with axonal vesicular trafficking and organelle transport, (2) reduced acetylation of the α-tubulin components of microtubules and consecutive impaired kinesin-1 binding and release of the molecular motor kinesin-1 from the microtubule rails, (3) abnormal phosphorylation of kinesin-1 which reduces its binding and interaction with the microtubule tracks, and (4) abnormal anterograde and retrograde trafficking of mitochondria (Fig. 8.2) (De Vos et al. 2008; Gunawardena and Goldstein 2005; Li and Conforti 2013; Millecamps and Julien 2013; Ortega et al. 2007).
8.2 The Neuronal Protein Quality Control Machinery: Molecular Chaperones and the Ubiquitin-Proteasome Pathway
Conformational changes and misfolding of specific disease proteins and their subsequent intraneuronal or extraneuronal aggregation are common molecular features shared by many neurodegenerative diseases including HD. Therefore, these human brain diseases are collectively referred to as proteinopathies, protein–misfolding disorders, or protein–conformational disorders (Atkin and Paulson 2014; Brundin et al. 2010; Goedert et al. 2010; Jucker and Walker 2011; Leak 2014; Ortega et al. 2007; Wooten et al. 2006). All types of intraneuronal and extraneuronal aggregations of conformationally changed and misfolded proteins which occur in human neurodegenerative diseases only develop within the framework of the ongoing disease process. They thus represent abnormal constituents of diseased brains. Since these aggregated, aberrant nerve cell constituents cannot be diluted by the postmitotic nerve cells through cell division, postmitotic neurons in particular rely on elaborated pathways of protein quality control and protein removal to prevent protein aggregation into pathological inclusions (Atkin and Paulson 2014; Ortega et al. 2007). Although the sequence of amino acids of a given protein contains all information necessary for its proper folding into a functional, three-dimensional structure, newly synthesized proteins may also undergo conformational change and misfolding, which together with a concomitantly decreased activity of the neuronal protein quality control machinery represents a risk factor for their intraneuronal accumulation and aggregation (Atkin and Paulson 2014; Brundin et al. 2010; Davies et al. 2007; Ortega et al. 2007; Paul 2008; Renner and Melki 2014; Wooten et al. 2006).
Related posts:
 Pathological Nerve Cell Alterations in Huntington’s Disease (HD) and Their Possible Role for the Demise of Nerve Cells
Conclusions and Outlook
Widespread Brainstem Neurodegeneration in Huntington’s Disease (HD)
Degeneration of Select Motor and Limbic Nuclei of the Thalamus in Huntington’s Disease (HD)
Consistent and Widespread Degeneration of the Cerebellum in Huntington’s Disease (HD)
The Neuropathological Grading of Huntington’s Disease (HD)
Pathological Nerve Cell Alterations in Huntington’s Disease (HD) and Their Possible Role for the Demise of Nerve Cells
Conclusions and Outlook
Widespread Brainstem Neurodegeneration in Huntington’s Disease (HD)
Degeneration of Select Motor and Limbic Nuclei of the Thalamus in Huntington’s Disease (HD)
Consistent and Widespread Degeneration of the Cerebellum in Huntington’s Disease (HD)
The Neuropathological Grading of Huntington’s Disease (HD)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


