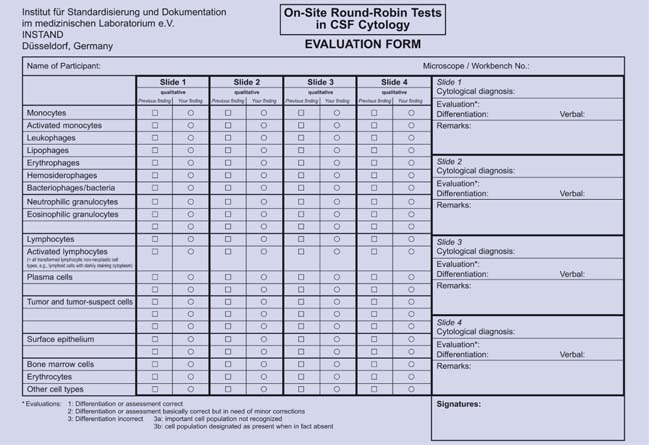
1 Introduction H. Kluge, V. Wieczorek, E. Linke, K. Zimmermann, O. W. Witte In 1954, in Jena, in what was then the German Democratic Republic (GDR), J. Sayk of the Hans Berger Clinic for Psychiatry and Neurology developed the cell sedimentation chamber that was later named after him, working in collaboration with the Carl Zeiss company. This was followed by a worldwide surge in cytological study of cerebrospinal fluid (CSF) for diagnostic purposes. The Sayk method of obtaining cells from the CSF, in which cells are normally rare, by spontaneous sedimentation was the least traumatic technique that had been developed up to that time, and indeed remains so today. Cytological examination of the CSF became a routine method of clinical laboratory investigation with the application of the panoptic Pappenheim stain already in use for blood and bone marrow cells (a combination of the May-Grünwald eosin–methylene blue stain with the Giemsa azure II–eosin stain) to the cells obtained by Sayk’s method. By the early 1960 s this technique, which originated in the Jena “school” of clinical CSF cytology of the late 1950 s (J. Sayk, V. Wieczorek, R. M. Schmidt), was adopted by centers in Rostock (J. Sayk when he took up the chair there, R. Olischer, H. Meyer-Rienecker, later R. Lehmitz) and Halle (R. M. Schmidt after assuming the chair), while continuing to be practiced in Jena (V. Wieczorek, later H. Kluge). These three centers soon initiated a program of training and continuing education in CSF cytology for heads of clinical and research laboratories and for technical assistants in CSF laboratories throughout the GDR; the laboratories themselves joined together to form a “Working Group for the Diagnostic Study of the Cerebrospinal Fluid and Clinical Neurochemistry.” The “Stadtroda Seminars in CSF Cytology,” which provide training and continuing education in the field, were founded in 1984 on the initiative of E. Linke, then head of the laboratory in the Stadtroda Academic Hospital (Fachkrankenhaus), and have taken place annually since then with the collaboration and supervision of V. Wieczorek (Jena) and the support of K. Zimmermann (Arnsdorf-Dresden), H. Kluge (Jena), and H. Krause (Berlin). The first nationwide German CSF symposium after the reunification of East and West Germany took place in Marburg in 1990 on the initiative of T. O. Kleine, E. Linke, and K. Zimmermann. In 1996, K. Felgenhauer, the G ttingen neurologist and President of the German Neurological Society, lent his support to the founding of the German Society for the Diagnostic Study of the Cerebrospinal Fluid and Clinical Neurochemistry, which is based in Jena. (We will refer to this Society by its German abbreviation, DGLN, which stands for Deutsche Gesellschaft für Liquordiagnostik und Klinische Neurochemie e.V.) In 1987 and 1988, E. Linke and K. Zimmermann conducted round-robin CSF cytology tests in Stadtroda. In each test, 12 CSF specimens were examined by teams from 22 participating CSF laboratories from across the GDR. These were, to our knowledge, the first such tests to be conducted in Europe. From 1995 onward, Linke and Zimmermann were able to conduct tests of this type on a nationwide scale in Germany. These events, known as “On-Site Round-Robin Tests for Quality Assurance in CSF Diagnostic Studies” (Ringversuche vor Ort zur Qualitätssicherung in der Liquordiagnostik), have been held annually ever since with the collaboration of the DGLN and the support of INSTAND, D sseldorf. They have already had a major, beneficial effect on the level of expertise in CSF cytology in Germany. Their most important benefit, however, has been the establishment of a uniform standard of cell classification. Over the years, many participants in these seminars and round-robin tests in CSF cytology expressed a desire for an atlas of practical CSF cytology for the purposes of training and continuing medical education, which would be compiled from the extensive collection of cytological specimens assembled over a period of about 40 years by V. Wieczorek and H. Kluge. The authors have attempted to meet this desire by putting together the present text, in which the major emphasis is placed, as always, on the initial morphological study of cells in the CSF in a May–Grünwald–Giemsa preparation. This will presumably remain the preferred initial method of cytological study of the CSF for some time to come, as it is the essential prerequisite for all further cytological studies and thus must be done first as a selective diagnostic filter. The cell picture obtained by this classical method often yields important diagnostic information; even if it does not, it leads the way to specifically targeted further studies that will be beneficial for both diagnostic and scientific purposes. Above all, this atlas is intended to provide an adequate grounding in CSF cytology for everyday practice in routine clinical laboratories and specialized CSF laboratories where such testing is done. The authors consider the need for such a text to be particularly pressing now that many of the previously independent CSF laboratories are being integrated into central laboratories whose technical and academic staff will need to be adequately trained in this subject. Chapters 2–6 will be especially useful for this purpose, because each of the numerous illustrations is accompanied by a brief discussion of the characteristic features and current World Health Organization (WHO) classification of the cell types depicted, and of the significance of the abnormal cytological findings in relation to the etiology and pathogenesis of the underlying disease. We would like to point out that detailed descriptions of cytological methods have been omitted in this atlas, as these can be found without difficulty in the relevant literature, which is extensive. Furthermore, we feel obliged to explain that, in order to illustrate some of the less common types of CSF pleocytosis encountered in rarer diseases, we have had no choice but to display older specimens of slightly lesser quality. Upto-date, advanced photographic techniques have been used to generate the illustrations in such cases and have greatly enhanced their appearance. For this, the authors are especially grateful to the staff of the Experimental Neurology Unit in the Hans Berger Clinics of Friedrich Schiller University, Jena, in particular to M. M. Kluska and C. Redecker, who expertly directed the photographic process using the clinical equipment of the Carl Zeiss company, Jena (Axioplan 2, AxioCam Hrc, AxioVision 3.1). We also thank Master Photographer W. Schumacher of Friedrich Schiller University for preparing the illustrations in timely fashion while the text was being written. Finally, we especially thank the technical staff of the CSF laboratory of the Jena Neurological Clinic for the preparation of the cytological specimens, in particular its long-serving Chief Technician, Ms. Adelheid Hoffmann. H. Kluge, V. Wieczorek, O. W. Witte, E. Linke, K. Zimmermann, E. Taub, S. Isenmann Classical cytological examination based on the Pappenheim stain (May–Grünwald–Giemsa stain) remains the centrally important first step in the cytological evaluation of the CSF, because it generally enables at least an initial categorization of the diagnosis into one of the major types of neurological disease. In some cases, it even allows further differential-diagnostic classification within a major disease category, thereby yielding much more useful information than a simple “yes” or “no.” However, classical cytological examination is rarely the final, conclusive test in differential diagnosis, because it usually fails to reveal the specific disease that is present, instead merely indicating the class of diseases to which it belongs. Yet this does not diminish its value as the point of departure from which the cytological diagnosis of the CSF will proceed. On the contrary, the information obtained by May–Grünwald–Giemsa staining is of crucial importance in determining whether other, more sensitive and more specific cytological marking techniques are necessary or superfluous for further evaluation. In many cases, decisions of this type can be made just on the basis of the clinical examination and the results of preliminary laboratory tests (e.g., findings in other areas of the body relevant to the patient’s illness, and/or validated analysis of humoral markers in the blood and CSF). In more concrete terms, classical CSF cytology is a rapid, inexpensive diagnostic technique that permits all of the following: 1. Detection of infectious diseases of the central nervous system (CNS) and their classification into major categories. Classical CSF cytological examination, when carried out either as an initial study or as a follow-up study in patients with CNS infections, yields information that determines which further test (s) should be done for identification of the pathogenic source (standard microbiological culture; enzyme-linked immunosorbent assay (ELISA) and blotting methods using pathogen-specific antibodies; molecular techniques, particularly hybridization/amplification with polymerase chain reaction (PCR), multiplex PCR, and blotting methods; immunocytochemical phenotyping of antibody-secreting cells, particularly with respect to immunoglobulin typing of activated B lymphocytes in the CSF). 2. Demonstration of acute, or older, hemorrhages into the CSF spaces that cannot be clearly revealed, or can no longer be clearly revealed, by neuroimaging studies (subarachnoid hemorrhage, hemorrhage accompanying inflammatory, traumatic, or neoplastic disease, etc.). Classical CSF cytological examination can also be used to distinguish true hemorrhage into the CSF space from the artifactual introduction of blood into the CSF. 3. Detection of neoplastic cells and their (at least initial) classification by origin in patients with an unknown or unclear oncological diagnosis (inadequate phenotyping of neoplastic cell populations of hematogenic origin, taken from the patient’s blood, bone marrow, or lymph nodes; probable primary CNS lymphoma; unknown primary tumor; and unclear cases in which true neoplastic cells must be distinguished from lymphocytic activated forms, pre-neoplastic stages, non-neoplastic progenitor cells, and non-neoplastic stimulated cell forms). In such cases, classical CSF cytology can be followed by appropriate immunocytological phenotyping techniques for cell-line–specific proteins (characterization of cell lines by the expression or nonexpression of epithelial, oncofetal, neuroendocrine, mesenchymal, neuron-associated, and gliaassociated antigens). 4. Follow-up studies and treatment monitoring in patients with pleocytosis and/or qualitative cellular changes in the CSF, as well as the monitoring of external ventricular drains for infection or inflammatory reaction to the catheter (material intolerance). The remaining sections of this chapter, and the text sections introducing the illustrations in later chapters, contain more detailed information about the indications, utility, and limitations of the special tests mentioned in points 1–4, above, in addition to classical CSF cytological examination. In addition, the cytological findings, together with the CSF cell count and protein concentration, may indicate the need for additional laboratory study of the noncellular components of the CSF (albumin quotient, intrathecal synthesis of various substances, humoral process markers, etc.). Cytological examination thus plays a key role in the integrated diagnostic evaluation of the CSF. Over the past decade in particular, we have noticed a marked increase in the number of specimens of ventricular CSF obtained via external ventricular drainage by neurosurgeons in our referral area. The most important clinical consideration here is obligatory monitoring for possible secondary catheter infections, which can occur at any time, despite optimal hygienic precautions, because of colonization of the ventricular catheter with pathogenic organisms. Two further uses of ventricular CSF cytology are the monitoring of reparative processes and the detection of incompatibility with the catheter material. The cytological examination of ventricular CSF with May–Grünwald–Giemsa staining is just as reliable as that of lumbar or cisternal CSF, because fluid obtained from all three of these areas has essentially the same cytological profile. The combination of cytological examination and standard microbiological culture of ventricular CSF constitutes an adequate battery of CSF tests when the purpose is to monitor for infection or for foreign-body reactions. The difference between the cytological picture of ventricular CSF and that of CSF from other sites is usually only quantitative: for certain structural and physiological reasons, surgical procedures and other disturbing factors in the ventricular system tend to produce a more marked granulocytic pleocytosis (neutrophilic and eosinophilic). This pleocytosis occurs in response to ventricular wall irritation, even in the absence of infection. In the authors’ experience, the ventricular CSF cell count often exceeds 500 cells/μL, with more than 75% neutrophilic granulocytes, even under broad-spectrum antibiotic coverage and in the absence of detectable microorganisms. In addition, macrophages of monocytic derivation are often found, as well as cells of epithelial/ependymal, mesenchymal, and endothelial origin and, occasionally, varieties of these cells that have been transformed into macrophages (cf. Chapter 4). The fundamental observations that we have made in this section and in the remaining sections of this chapter, as well as in the introductory text preceding the illustrations of special cytological findings in the other chapters of this book, are based not only on our own experience but also on information that can be found in relevant chapters and literature cited in other specialized textbooks in the field. Some of these texts are fairly recent (Thomas 1998; Felgenhauer and Beuche 1999; Zettl et al. 2003, 2005), whereas others are older (Kölmel 1978; Dommasch and Mertens 1980; Schmidt 1987). Likewise, to complement our discussion of the cytological findings with the indispensable further information from other clinical neurosciences, we have made use of the basic literature in neuropathology (Cervos-Navarro and Ferszt 1989) and of the WHO classifications of tumors of the nervous system (Kleihues and Cavenee 2000; Radner et al. 2002). A number of cytological atlases of the CSF have been published, but none are recent: Oehmichen (1976), Kölmel (1978), Schmidt (1978), Den Hartog Jager (1980). A few of the major recent findings in basic science are mentioned in this book in the appropriate places. Finally, there is a list of older works at the end of the book, relating to the subject matter of Chapters 3–5, which were important milestones in the development of the Jena school of CSF cytology (Sayk et al.). M. Wick, H. Kluge, E. Schulze, R. Lehmitz, M. M. Kluska In the classical cytologic examination of the CSF, cell preparation with the May–Grünwald–Giemsa stain usually permits the basic identification and a fairly precise classification of malignant and inflammatory cell populations. Cells of these two types may be difficult to distinguish from one another, however, and subclassification is often not possible. Thus, the additional use of immunocytological phenotyping techniques may be necessary. When deciding whether to use such techniques, the CSF diagnostician must first ask whether the potential findings will be helpful for diagnosis and treatment, i.e., whether these techniques will really provide any clinical benefit. For example, if myelogenous or lymphocytic neoplastic cell populations or immature preneoplastic stages have already been detected in specimens of blood, bone marrow, or lymph nodes, so that the diagnosis of myelogenous or lymphocytic leukemia or lymphoma has already been made or ruled out, additional immunocytological phenotyping of the CSF cellular sediment is unnecessary. The same holds for brain metastases if the primary tumor is already known. Thus, immunocytological phenotyping of CSF cells is indicated only for the classification of atypical cells when a tumor of uncertain origin or primary CNS lymphoma is suspected (“tumor-suspect” cells on May–Grünwald–Giemsa staining, particularly when only a few cells are seen), or for the differentiation of lymphocytic-inflammatory from lymphocytic-neoplastic cell populations. Immunocytological phenotyping is recommended, although it is not indispensable, for the quantification of subclasses of activated B lymphocytes (B cells), particularly in chronic inflammatory diseases, if the May–Grünwald–Giemsa preparation reveals only a few activated lymphocytes, or only mildly stimulated ones. (For further details, see Chapter 3, Cytological Findings in Infectious and Inflammatory Diseases and the corresponding chapters in Zettl et al. 2003, 2005.) The antibodies used for phenotyping are the same for all techniques regardless of the tissue being studied, depending only on the particular cell lineage that is to be demonstrated. They can be used for the study of CSF cells just as well as other tissues. A major limiting factor for the immunocytological phenotyping of CSF cells is the amount of cellular sediment that is available for study. This amount determines the technique that must be used. Each immunocytological marking requires 1000–10 000 “white cells,” depending on the frequency of the specific cell population being sought; thus, when the CSF contains only rare cells, flow cytometry is difficult or impossible to perform, even with the use of double or triple antibody marking (see Diagnostic Use of Flow Cytometry in CSF Cytological Examination below). It can be done only if a large volume of CSF is centrifuged to increase the cellular concentration before cytometric testing. Microscopic techniques for the phenotyping of cellular sediment (immunofluorescence microscopy and immunocytochemical techniques) require at least 1000 and preferably at least 5000 cells per marking. These techniques are carried out after cellular enrichment by sedimentation and provide usable results even when the CSF contains only rare, atypical cells of the type (s) to be demonstrated. However, multiple antibody marking is then difficult and, therefore, a larger number of sedimented specimens is required. (Our experience confirms this principle, especially in the case of “tumor-suspect” cells.) If “tumor-suspect” and other atypical cells are seen in the May–Grünwald–Giemsa preparation and the CSF laboratory does not have the on-site capability for immunocytological phenotyping, or the necessary experience for it, we recommend that some of the excess CSF not needed for other tests be used to prepare more slides of air-dried, unstained sediment (the lower the total cell count, the more such slides should be made), and that these slides then be speedily sent to a specialized laboratory for phenotyping. A comprehensive discussion of critical points concerning analytical evaluation, differential diagnostic clusters, and the corresponding antibody panels can be found in Thomas (1998) and Zettl et al. (2003, 2005). E. Sindern, M. Wick, M. Roskos, M. Kiehntopf, H. Kluge For the study of body fluids with a high concentration of cells, such as blood, flow cytometry is now well established as a routine diagnostic technique that has clear advantages over the traditional microscopic methods. Its potential application to the study of CSF is more limited, however, because normal CSF is poor in cells, with a white cell concentration generally no more than 1/1000 of that of blood. Whenever flow cytometric study of the CSF is both technically feasible and clinically indicated, it is recommended that it should be done in combination with a classical differential cytological examination of the CSF in a May–Grünwald–Giemsa preparation. The reasons for these general considerations are given below. First, the flow cytometers now routinely used for hematology and urinalysis (ADVIA 120, CellDyn 3500 and 400, Sysmex UF-100) measure the cellular parameters of size, volume, surface area, granularity, and fluorescence with the use of two fluorescent dyes, phenanthridine for DNA and carbocyanin for cell membranes. They can be used to determine the total cell count of the CSF, as well as a differential cell count that includes erythrocytes (red blood cells [RBCs]), mononuclear cells (lymphocytes and monocytes), and polymorpho-nuclear cells (neutrophilic and eosinophilic granulocytes). The mononuclear cell count, however, includes only normal lymphocytes and monocytes. Pathological mononuclear cell populations, whose detection is often of crucial importance to neurological diagnosis, have extremely varied characteristics with respect to the analytical parameters measured by these cytometers. Activated lymphocytes (see Chapter 3), activated monocytes (including macrophages, see Chapter 4), many different types of tumor cell and tumor-suspect cell (including mitoses, see Chapter 5), bone marrow components, and epithelial cells (see Chapter 2) remain undetected by these cytometers, or else they are mistakenly assigned to one of the three categories mentioned. Therefore, no experienced CSF cytologist or responsible clinical neurologist can share the highly optimistic assessment of Aune et al. (2004), who consider a “CSF assay” with the ADVIA 120 apparatus to be an acceptable alternative to manual microscopic methods. One must rather agree with the more critical opinion of Strik et al. (2005), who consider the use of this apparatus acceptable only for emergency testing, and even then only in combination with a simultaneous cell preparation for microscopic analysis. As responsible clinical neurologists Strik et al. take a critical and objective view of the methodological and diagnostic limitations of flow cytometry. Second, CSF flow cytometry coupled with immunocytological characterization of diagnostically relevant cell populations can certainly be of major diagnostic importance. In the remainder of this section, we will describe the technical principles and sphere of application of these techniques. The following conditions and details must be taken into consideration. When the CSF specimen contains an adequate number of cells for analysis the clinical benefits of flow cytometry can be fully realized. Functionally distinct cell populations that are difficult or impossible to differentiate from one another with classical staining techniques can often be distinguished by flow cytometry based on varying amount of binding of fluorescence-marked monoclonal antibodies to their surface and intracellular antigens, because different cell populations bear antigens of different types and/or density (immunological phenotyping, cf. Diagnostic Use of Immunocytological Phenotyping Techniques in CSF Cytology). Furthermore, flow cytometry enables examination of all of the nucleated cells within the fluid volume being studied, and not just with a single antibody to a single antigen (a limitation of routinely used light microscopy) but with simultaneous combinations of antibodies, so-called “antibody panels,” directed against two or three different antigens (multiparametric analysis). It provides a quantitative measure of the fluorescent activity of the labeled cells. The ability to examine a large number of cells at once enables detection of sparser cell populations than can be detected by microscopic techniques. Flow cytometry is a rapid, highly automated technique with steadily expanding uses for the examination of many different body fluids that contain cells. It has already been used for cell-kinetic and functional studies (enzyme activity, receptors, cell proliferation and cell cycles, inflammatory mediators, apoptosis, etc.). However, flow cytometry, coupled with immuno-logical characterization is difficult or impossible to carry out on CSF samples with a normal or only mildly elevated cell count. Each lumbar puncture yields an intrinsically limited quantity of CSF, lumbar punctures cannot be repeated ad libitum, and much of the CSF that is obtained must be sent for routine cytological, microbiological, and clinical chemical studies, so that, finally, little or none will remain for flow cytometry (a possible need for cell enrichment by centrifugation will only worsen the problem). These difficulties can sometimes be circumvented by sending only the acellular component of the centrifuged CSF for clinical chemistry; indeed, testing of this component for humoral markers even suffices to establish the diagnosis in some cases, obviating the need for flow cytometry. If enrichment by centrifugation is necessary to obtain enough cells for all of the antibody (or antibody panel) tests that are to be done, including negative controls, one must expect a loss of about 40% of the cells through the process of centrifugation—up to 30% of lymphocytes and granulocytes and up to 80% of monocytes. Furthermore, as flow cytometry involves the statistical sampling of cell populations, its results generally have a binomial distribution: this implies that, if only a few cells are available for analysis, the result will lie within a wide confidence interval, i.e., the information obtained by flow cytometry will be relatively imprecise. The coefficient of variation for the repeated analysis of a single cell population has been reported to be as high as 30% (depending on the population studied). How many cells are needed for analysis with currently available equipment, and what are the current applications of flow cytometry in the examination of the CSF? In response to the first question, the number varies depending on the percentage of all CSF cells that is represented by the particular cell population to be studied, out of which at least 100–200 cells should be present to ensure a reliable analysis. This general principle allows a critical assessment of statements in the literature on threshold levels for the total number of white cells. According to Dux et al. (1994), the analysis of lymphocyte subpopulations with the FACS-II System requires at least 1000 CSF cells per antibody panel (account has already been taken of 30–40% cell loss through centrifugation). With a normal cell count of 1–5 cells/μL, this corresponds to a required CSF volume of approximately 2 mL for each antibody panel (including negative control). On the other hand, if a viral inflammatory process has elevated the CSF cell count to 200 cells/μL a 50μL aliquot would already contain 10 000 cells and could be used for flow cytometry directly, without centrifugation. Sindern’s laboratory, using the same apparatus, requires at least 2000 cells per antibody panel, including negative control. Kleine et al. (1994) report the ability to analyze eight lymphocytic subpopulations with a total of 3–5 mL of CSF in aliquots of 100 μL of native CSF, as long as the cell count exceeds 10/μL (yielding at least 1000 cells per aliquot). Wick (in Zettl et al. 2003, 2005) states that at least 10 000 cells are needed for immune phenotyping of rarer types of lymphocytes. The number of cells needed per antibody panel thus varies from 1000 to 10 000. If these cells are to be obtained from an aliquot of 50 mL of native CSF, then the CSF cell count must be at least 20/μL (at least 100/μL is desirable). These quantitative requirements, the other disadvantages of flow cytometry mentioned above, and the fact that it can only be used to study a small number of cell populations per test limit the current clinical applicability of flow cytometry to the cytologic analysis of CSF. Indeed, CSF flow cytometry is routinely done in only a few clinical laboratories and is more commonly used as a research tool. It is usually used to study lymphocytic cell populations, as lymphocyte loss through centrifugation (up to 30%) is acceptably low and seems to affect all subpopulations equally. Studies of lymphocytic pleocytosis have revealed a compartmentalization of lymphocytic subpopulations between the blood and the CSF: the CSF contains a significantly higher percentage of CD3+ cells (T lymphocytes), CD4+ cells (helper T lymphocytes), and CD4+ CD29+ cells (activated helper-memory T lymphocytes) than the blood, which, in turn, contains a higher percentage of CD19+ cells (B lymphocytes), CD8+ cells (suppressor T lymphocytes), and CD4+ CD45RA+ cells (naïve helper T lymphocytes). These differences are independent of the underlying condition. CSF of patients with relapsing-remitting multiple sclerosis (MS) has been found to contain a significantly higher percentage of CD19+ and CD4+ CD29+ cells than that of patients with viral meningitis, viral encephalitis, or other, noninflammatory diseases of the nervous system. Most CD3+ cells in the CSF express the chemokine receptor CXCR3; in patients with MS, the degree of CXCR3 expression correlates with the extent of active inflammation revealed by magnetic resonance imaging (MRI) and perhaps reflects preferential recruitment of activated cells from the blood into the CSF (Oreja-Guevara et al. 1998, Sindern et al. 2002, Sindern 2004). These findings, however, cannot yet be put to clinical use, nor does our current knowledge of the different lymphocytic composition of the CSF in various neurological diseases (mostly infectious/inflammatory) have any immediate clinical relevance beyond its usefulness in answering specific scientific questions. There is a potential future clinical application for CSF flow cytometry in the differential diagnosis of neoplastic meningitis. Further information on CSF flow cytometry can be found in the relevant chapters in the cited literature (Thomas 1998; Zettl et al. 2003, 2005). H. Kluge, M. Roskos, E. Linke, V. Wieczorek, E. Taub, S. Isenmann The importance of proper handling of CSF specimens before cytological examination is easily understood as soon as one grasps the single most important reason for cell damage after lumbar puncture. The CSF normally contains little protein and only a few cells and thus mostly lacks the two main pH buffer systems that are present in blood, namely cell surfaces and a high protein concentration. Practically the sole buffering capacity of the CSF is derived from the bicarbonate/carbon dioxide system. A drop of CSF suspended from the lumbar puncture needle gives up its carbon dioxide content to the surrounding air because of the steep partial pressure gradient for carbon dioxide and thereby spontaneously loses the only pH buffering capacity that it has. Within seconds, therefore, the pH rises from 7.32–7.36 (normal CSF range) to alkaline values of 7.8 and higher. If the CSF is not very bloody (as in most normal and pathological specimens) and the CSF protein concentration is below 3000 mg/L, then, regardless of the white cell count, the elevated pH in combination with active catabolic mechanisms will cause a considerable degree of cell damage and cell loss even at this early stage. (The old aphorism about the CSF being a “hostile environment for cells” is misleading, as this is true only of CSF exposed to air.) On the other hand, bloody CSF with a high protein content contains adequate buffers, i.e., pH-stabilizing ampholytes on erythrocyte surfaces and protein molecules, so that the pH does not rise as rapidly and cell damage does not occur till much later. Experienced cytologists are well acquainted with this phenomenon and can obtain a diagnostically useful preparation and differential cell count from a bloody specimen as long as 4–6 hours after lumbar puncture. After a longer interval this is no longer possible because of cell loss and artifact formation. The method of cell sedimentation is another important factor affecting cell stability (for details see below). Spontaneous sedimentation, e.g., in the Sayk chamber, is less damaging than cellular centrifugation, whose harmful effect is increased if the cells have already been damaged previously by medical treatment or by having been left standing too long after the lumbar puncture. The strict observance of a time limit (see below) is thus all the more important nowadays, when cell sedimentation is almost always done with a cell centrifuge. The storage temperature of CSF should be between 5 ° C and 12 ° C to minimize cell damage. Lower temperatures may lead to cold lysis, whereas higher temperatures accelerate the catabolic mechanisms mentioned above. The best receptacles and transport containers for CSF are sterile, sealed polypropylene test tubes. Polycarbonate test tubes adsorb various substances to their surface, e.g., IgG, whereas glass test tubes should not be used because they adsorb cells and therefore artifactually alter the cell count. EDTA and sodium fluoride test tubes also impair cytological examination. Keeping to a time limit from the moment of the lumbar puncture until the cytological examination is important not only because of progressive cell damage, but also because any cells present (particularly in CSF pleocytosis) can damage, or bind to, sensitive noncellular substances used as diagnostic markers, thereby preventing their detection. Aliquots of CSF that are to be used for diagnostic marker studies should have all cells removed as soon as the CSF specimen arrives in the laboratory. Taking into account all of the mechanisms that can damage cells, it can be concluded that the cytological examination of the CSF should take place no later than 2 hours after lumbar puncture. Experienced CSF cytologists can provide useful diagnostic information even after a time interval of 3 hours, but at 4 hours any findings must be accompanied by statement of caution. For all these reasons, central cytological laboratories that process specimens from a wide referral area (and, of course, central laboratories that perform cytologic examination along with other tests) must establish a strict set of rules for specimen handling, both externally (at the referring hospital and in the transport system) and within the laboratory (assignment of specimens for processing, maintenance of technical standards). These rules should be adapted to local conditions. Referring physicians, in particular, must realize that lumbar punctures cannot be repeated at will, especially because of patient discomfort and the likelihood of postpuncture reactions, and therefore specimens must be carefully handled from the moment of lumbar puncture until they arrive in the laboratory. There are other considerations that the physician should be aware of when taking specimens of ventricular CSF from external drainage systems. The (approximately) 2 mL of CSF lying between the catheter tip and the point of removal of specimens may have undergone secondary changes, as this portion of fluid has been lying outside the ventricular system for some time (the length of time depends on the volume of drainage per hour as well as on the diameter and length of the drainage tubing). This should be removed and discarded if possible. Other recommendations for the removal of ventricular CSF specimens can be found in the relevant chapters in Zettl et al. (2003, 2005). The basic cytological examination (cell count, differential cell picture, and total protein) requires a minimum of 2 mL of CSF, though the precise amount may vary between laboratories. More CSF is needed if the suspected diagnosis calls for further specialized cytological testing (immunocytochemistry or flow cytometry). In such cases, the referring physician should contact the CSF laboratory for advice before carrying out the lumbar puncture. The same holds if tests are to be performed on solutes both in the acellular component of CSF and in the autologous serum (quotients of albumin, immunoglobulins, etc.). Our daily experience shows it is essential for CSF laboratories to provide detailed information to all referring institutions (departments of neurology, neurosurgery, anesthesiology, pediatrics, and psychiatry; medical oncology wards; etc.) about the proper handling of specimens and the required minimum volumes of CSF and autologous serum (if the serum is to be tested). H. Kluge, M. Roskos, M. M. Kluska The preparation of the cell sediment is among the more difficult technical components of the cell enrichment process because it is subject to a number of potential disturbances. The Sayk method of spontaneous cell sedimentation, which is mainly used to obtain populations of easily damaged, highly activated large-sized cells, is today still the best method for the preservation of cellular structure, superior in this respect to all methods involving filtration or centrifugation. The literature contains many quantitative and qualitative comparisons of these methods; here it will suffice to mention the representative and comprehensive paper by Lehmitz and Kleine (1994). Nonetheless, improvements in centrifugation techniques over the years have made some of these techniques nearly as good at preserving cell structure as spontaneous sedimentation, with the further advantages of rapidity and constancy of technical parameters. Standardized centrifugation techniques are now in wide use; the most popular cell centrifuges are those manufactured by Shandon Labortechnik (Frankfurt, Germany) and Hettich (Tuttlingen, Germany). Among the cell preparations shown in this cytological atlas, those produced before 1990 (the majority) were all made with Sayk’s method of spontaneous cell sedimentation. From 1990 until about 1993, the authors made parallel preparations with the Sayk method and with cell centrifugation techniques, in an effort to raise the quality of the latter to the standard set by the former by modifying the centrifugation parameters and adding certain cell-protective substances. In these trials, we were indeed able to produce centrifuged cell preparations of acceptably similar quality to those made with the Sayk method. Yet we must stress that the centrifugation technique used by us and described further below, like any other centrifugation technique, cannot be applied in rote, unvarying fashion to every single specimen; rather, it must be optimally adapted to each specimen to obtain the best result. This is particularly true when an initial preparation has already been made and turns out to be of poor quality. The technical parameters that we give here for our “Jena version” of the technique should therefore be understood as rough guidelines derived from experience, allowing room for flexible application. The particular variant of the cell centrifugation technique that we developed employs the UNIVERSAL 32 cell centrifuge with an angle chamber system (manufactured by Hettich, Tuttlingen, Germany) and is based on one-step centrifugation without any preliminary centrifugation enrichment of the type previously used for CSF specimens with a low cell count (two-step centrifugation). If the specimens have been handled properly before analysis (proper transport, keeping to time limits, etc., see above), this one-step technique yields diagnostically useful results without the cell loss and possible cell deformation caused by preliminary centrifugation. The technical details are as follows: Among the centrifugation programs that we tested, the following were found to produce an acceptably low amount of cell damage: 100 × g (930 rpm with the above-mentioned centrifuge) for 3 minutes or 50 × g (660 rpm) for 4 minutes. The former is recommended for routine use. The cell yield for CSF specimens with cell counts under 3/μL was 25–35%. Our deliberate use of a relatively low centrifugal acceleration to avoid cell damage explains why this cell yield is lower than that obtained by Lehmitz and Kleine (1994) in a two-step process using vertical cell chambers and an acceleration of 250 × g for 3 minutes. We generally used a standardized final fluid volume of 450 μL per angle chamber. Out of this volume, 400 μL consisted of CSF and a variable amount of diluent depending on the cell count; the remaining 50 μL consisted of the required additives described below. A simple calculation shows that 400 mL of CSF with a cell count of 2/μL and a 25% cell yield will provide 200 cells, an adequate number for cytologic analysis. If there are fewer than 2 cells/μL, a higher CSF volume of up to 800 mL can be used, as this amount of fluid can still be taken up by the filter card. If the cell count is higher than 2/μL, a smaller quantity of CSF is used, together with the additives mentioned below for a final volume of 400 μL. The procedure outlined above utilizes angle chambers of 8.7 mm diameter (sedimentation surface area 60 mm2), the corresponding filter cards, and Polysine-coated object carriers (for the May–Grünwald–Giemsa stain) or Super Frost Color object carriers (for immunocytochemical stains). Protein additives are needed for pH buffering and cell stabilization if the CSF cell count is below 10/μL and its protein content is below 3000 mg/L (see Proper Handling of CSF Specimens Before Cytological Examination). The protein concentration can always be raised well above 3000 mg/L, if necessary, by adding 50μL of freshly obtained and re-centrifuged normal serum (which is readily available in all laboratories and contains about 70 g/L of total protein) to the 400-μL specimen. (There is no need to use autologous serum.) The protein concentration of a specimen treated in this way will be approximately 7800 mg/L plus the protein concentration of the original CSF specimen. Antibiotics and protein-rich culture media were used at one time as additives to CSF for centrifugation, but these substances are both expensive and of no benefit at all, as the cytological preparation must be accomplished rapidly in any case if it is to be diagnostically useful. Bloody CSF with many cells should be diluted by mixing an aliquot of CSF of the appropriate size with a 10:1 mixture of normal saline and serum. The air-dried preparation is then stained in standard fashion for CSF sediment, with May-Grünwald solution for 2–3 minutes, then with Giemsa solution for 10–15 minutes. These times should be taken as guidelines and not as dogma. The creation of a standardized reporting form for CSF cytology was discussed in 1991 at the founding meeting of the (German) Working Group for the Diagnostic Study of the Cerebrospinal Fluid and Clinical Neuro-chemistry. The chief participants were neurologists and heads of CSF laboratories who were all well acquainted with the diagnostic importance of CSF cytology in clinical practice and who had themselves published important work in this area (Sayk, Wieczorek, Olischer; Schmidt and Kölmel; authors of earlier cytologic atlases of the CSF; and others). The Working Group produced a consensus reporting form, which is reproduced in English in Figure 1.1. The consensus reporting form has been used since 1995 in the nationwide on-site round-robin tests that first began in the Stadtroda CSF laboratory (E. Linke and K. Zimmermann, cf. Role of Practical (Classical) CSF Cytological Examination and the chapter devoted to this subject in Zettl et al. 2003, 2005). Figure 1.1 shows the current version, which can be used to record not only the cytological findings (cell populations present and absent) but also the evaluating cytologist’s judgment of the findings of the previous examiner. All participants agreed at the outset that the form should have spaces to indicate the presence or absence of monocytes, activated monocytes, phage and granulocyte subpopulations, and lymphocytes. There was considerable discussion, however, about the many subtypes of lymphoid cells that are seen in May–Grünwald–Giemsa preparations, and their many transitional forms, which are nearly impossible to classify precisely and reproducibly. The presence of these immunocompetent subpopulations in the CSF is indeed evidence of an inflammatory reaction, but it does not imply anything else about the differential diagnostic classification; therefore, it was finally agreed that all these cell types would be designated by the collective term “activated lymphocytes,” which is meant to encompass all transformed, non-neoplastic lymphocytic cell types. A distinct category was maintained only for plasma cells, which can be considered the last transformational stage of the lymphocytic series—even though subtypes of plasma cells, too, are sometimes difficult to distinguish from activated lymphocytes in classical cytological preparations. The latter problem is discussed in greater detail in Chapter 3. The differential diagnostic classification of lymphocytic subpopulations is possible only with immunocytochemical techniques, as discussed above and in the relevant chapters of Zettl et al. (2003, 2005). The participants also had varying ideas about the classification of neoplastic cell populations. It was finally agreed that there would be a space to indicate the presence or absence of neoplastic and tumor-suspect cells, as well as separate blank lines where the examiner could further specify the type of malignant cell in cases of suspected lymphoma or leukemia, if desired. The extensive presentation of neoplastic cell populations in Chapter 5 of this book contains the type of information that is to be specified on these lines in relevant cases. The term surface epithelium, discussed further in Chapter 2 (see Cells and Cell Clusters From the Structures Enclosing the CSF Space), refers to the cells and cellular networks that form the boundaries of the CSF space (subarachnoid space). The need to include bone marrow cells on the reporting form is justified in Chapter 2 (see Lumbar Puncture Artifacts). An indication of the possible presence of erythrocytes, due to a real or artifactual admixture of blood, is clearly indispensable as well. Finally, the last line is reserved for mention of other cell types, if present.
Role of Practical (Classical) CSF Cytological Examination in the Overall Spectrum of CSF Diagnostic Studies
Diagnostic Use of Immunocytological Phenotyping Techniques in CSF Cytology
Diagnostic Use of Flow Cytometry in CSF Cytology
Proper Handling of CSF Specimens Before Cytological Examination
Cell Preparation (Sedimentation) and Staining
1. Centrifugation parameters for cell sedimentation and CSF volumes to be used
2. Accessories, additives, and staining
The CSF Cytology Report: Consensus Reporting Form of the German Society for the Diagnostic Study of the Cerebrospinal Fluid and Clinical Neurochemistry for Use in On-Site Round-Robin Tests
< div class='tao-gold-member'>
Related posts:
 Pathological CSF Cell Findings in Cysts
Pathological CSF Cell Findings in Cysts
 Cell Populations in the Normal Cerebrospinal Fluid
Cell Populations in the Normal Cerebrospinal Fluid
 Pathological CSF Cell Findings in Infectious and Inflammatory Diseases of the Central Nervous System
Pathological CSF Cell Findings in Infectious and Inflammatory Diseases of the Central Nervous System
 Pathological CSF Cell Findings in Intracranial Hemorrhage and Traumatic and Hypoxic-Ischemic Brain Injury
Pathological CSF Cell Findings in Intracranial Hemorrhage and Traumatic and Hypoxic-Ischemic Brain Injury
 Pathological CSF Cell Findings in Primary and Metastatic CNS Tumors, Malignant Lymphoma, and Leukemia
Pathological CSF Cell Findings in Primary and Metastatic CNS Tumors, Malignant Lymphoma, and Leukemia
 4: Isolated Sleep Paralysis: An REM-“Sleep” Polysomnographic Phenomenon as Documented With Simultaneous Clinical and Electrophysiological Assessment
4: Isolated Sleep Paralysis: An REM-“Sleep” Polysomnographic Phenomenon as Documented With Simultaneous Clinical and Electrophysiological Assessment



