, Jean Paul G. Vonsattel2, Helmut Heinsen3, 4 and Horst-Werner Korf5
(1)
Dr. Senckenbergisches Chronomedizinisches Institut, Goethe University Frankfurt, Frankfurt, Germany
(2)
Medical Center Neurological Institute, Columbia University, New York, NY, USA
(3)
Division Psychiatic Clinic Morphological Brain Research Unit, Julius Maximilians University Würzburg, Würzburg, Germany
(4)
University of Sao Paulo Medical School, Sao Paulo, Brazil
(5)
Dr. Senckenbergisches Chronomedizinisches Institut, Goethe University, Frankfurt, Frankfurt, Germany
Since the late 1880s hereditary chorea is named Huntington’s disease after George Huntington (1850–1916) (Fig. 1.1) who provided the classical description of adult onset of hereditary chorea in his essay “On chorea” from 1872. However, this publication was not the first clinical description of adult-onset hereditary chorea. It was preceded by earlier and fairly complete clinical descriptions (e.g., that of Waters, Lund, or Lyon) which already mentioned many of the key clinical features of the disease including its hereditary nature, insidious onset in adulthood, and progressive disease course (Heathfield 1973; Huntington 1872; Lanska 2000, 2010; Lund 1860; Lyon 1863; Waters 1842). In his essay from 1872, Huntington incorporated the medical records of the patients treated previously by his father and grandfather and noted the hereditary transmission of chorea, its gradual onset during adulthood and progressive course, the tendency of affected patients to insanity and suicide, as well as the resistance of the disease to treatments. In its main part this historical essay dealt with rheumatic or Sydenham’s chorea, but its last part was devoted to a hereditary chorea with mental changes commencing in midlife, which is transmitted from generation to generation and follows a remorselessly progressive course (Heathfield 1973; Huntington 1872; Lanska 2000, 2010).

Fig. 1.1
The physician George Huntington (Reprinted from Lanska (2000), (Figure 2, page 83); with kind permission from Taylor & Francis)
Widely believed to represent the initial descriptions of the brain pathology associated with Huntington’s chorea and constituting milestones in neuropathological HD research, the neuropathological reports of Gerbrandus Jelgersma and Alois Alzheimer by no means constitute the first descriptions of the brain pathology in HD (Alzheimer 1911; Heathfield 1973; Jelgersma 1908; Lange and Aulich 1986).
From today’s point of view, many of the historical neuropathological observations and descriptions revealed very valuable and pioneering postmortem findings. Although widely forgotten or neglected, many findings and assumptions of these early postmortem HD studies have been repeatedly confirmed during the following period of more than hundred years of neuropathological HD research. Certainly, the short historical review provided in the introduction of this monograph cannot do justice to all the involved HD researchers of the past, but it tries to capture the essential milestones of neuropathological HD research, to describe them in detail, and to explicitly appreciate their importance for the current neuropathological knowledge in the field of HD and its stepwise development during the last century. Owing to the large amount of neuropathological investigations performed worldwide during the last century, it is impossible to describe all findings of all these previous postmortem studies in this monograph in detail. Therefore, we have restricted our efforts to a collection of the most important discoveries of neuropathological HD research. With regard to the assumptions and confirmed, questionable, and unconfirmed postmortem findings of previous HD studies not mentioned here, we must refer to the original neuropathological literature.
Ewald Stier possibly was among the first neuroscientists who pointed to widespread neurodegenerative findings in the brains of patients with Huntington’s chorea, which, astonishingly, are already compatible with the current view of HD as a multisystem neurodegenerative disease. Along with macroscopical brain alterations (i.e., reduced brain weight and brain atrophy), extensive degenerative changes were reported in subcortical brain regions (e.g., thalamus, putamen, and pallidum) as well as in circumscribed regions of the cerebral cortex (Stier 1903). In 1908, Gerbrandus Jelgersma described atrophic changes of the striatum (i.e., caudate nucleus and putamen), pallidum, and cerebral cortex in a patient with chronic chorea and attributed the occurrence of involuntary choreatic and incoordinated movements to these degenerative alterations in the striatum (see Chaps. 2, 3, and 4) (Jelgersma 1908).
Alois Alzheimer (Fig. 1.2) apparently also recognized the widespread brain pathology in Huntington’s chorea in 1911. Along with degeneration of the striatum and pallidum, he mentioned the involvement of additional subcortical regions (e.g., thalamus, brainstem, and spinal cord) and the cerebral cortex and attributed the occurrence of chorea to the degeneration of the striatum (see Chaps. 2, 3, 4, and 7). Currently representing a well-known and universally accepted, characteristic histopathologic feature of HD, the differential vulnerability of small and large striatal nerve cells for the degenerative process of HD has been delineated by James Ramsey Hunt and among others has been confirmed by Cécile and Oskar Vogt in their classical, comprehensive work on the human striatum and its diseases (see Chaps. 2 and 4) (Hunt 1917; Vogt and Vogt 1920). In their seminal work Cécile and Oskar Vogt (Fig. 1.3) mentioned, apart from degeneration of the striatum and pallidum, an involvement of the claustrum, subthalamic nucleus, and spinal cord in chorea patients, as well as a widespread loss of cortical pyramidal cells and preservation of the giant Betz pyramidal cells in the primary motor cortex (see Chaps. 2, 3, 4, and 11) (Vogt and Vogt 1920). Subsequently Friedrich H. Lewy described an additional affection of the substantia nigra and cerebellum in chorea patients (see Chaps. 5, 6, and 7) (Lewy 1923). Early reports of a possible involvement of gray components of the medial temporal lobe in chorea patients are scanty and pointed to degenerative changes in the allocortical hippocampus (Terplan 1924) and amygdala (see Chaps. 3 and 11) (Davison et al. 1932).
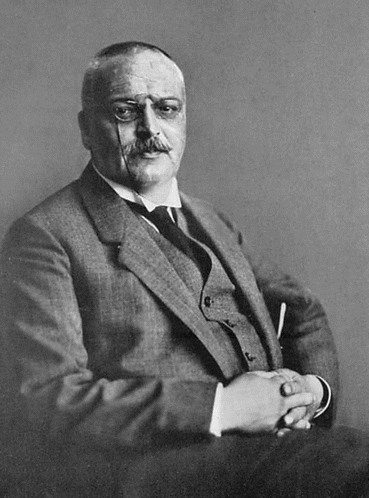
 Pathological Nerve Cell Alterations in Huntington’s Disease (HD) and Their Possible Role for the Demise of Nerve Cells
Conclusions and Outlook
Pathological Nerve Cell Alterations in Huntington’s Disease (HD) and Their Possible Role for the Demise of Nerve Cells
Conclusions and Outlook
 Widespread Brainstem Neurodegeneration in Huntington’s Disease (HD)
Widespread Brainstem Neurodegeneration in Huntington’s Disease (HD)
 Intraneuronal Transport and Defense Mechanisms with Possible Pathogenetic Relevance in Huntington’s Disease (HD)
Intraneuronal Transport and Defense Mechanisms with Possible Pathogenetic Relevance in Huntington’s Disease (HD)
 Degeneration of Select Motor and Limbic Nuclei of the Thalamus in Huntington’s Disease (HD)
Degeneration of Select Motor and Limbic Nuclei of the Thalamus in Huntington’s Disease (HD)
 Consistent and Widespread Degeneration of the Cerebellum in Huntington’s Disease (HD)
Consistent and Widespread Degeneration of the Cerebellum in Huntington’s Disease (HD)




Related posts:
Pathological Nerve Cell Alterations in Huntington’s Disease (HD) and Their Possible Role for the Demise of Nerve Cells
Conclusions and Outlook
Widespread Brainstem Neurodegeneration in Huntington’s Disease (HD)
Intraneuronal Transport and Defense Mechanisms with Possible Pathogenetic Relevance in Huntington’s Disease (HD)
Degeneration of Select Motor and Limbic Nuclei of the Thalamus in Huntington’s Disease (HD)
Consistent and Widespread Degeneration of the Cerebellum in Huntington’s Disease (HD)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
