Axis 1: Ictal phenomenology. Description of Ictal events using the Glossary of Descriptive Ictal Terminology (Blume, 1991)
Axis 2: Seizure type. Specifying localization within the brain and precipitating stimuli for reflex seizures using the List of Epileptic Seizures
Axis 3: Syndrome. Specifying possible syndrome from the List of Epilepsy Syndromes. A syndromic diagnosis may not always be possible though
Axis 4: Aetiology. Defining when is possible genetic defects and pathologic underlying causes for symptomatic focal epilepsies from the Classification of Diseases Frequently Associated with Epileptic Seizures or Epilepsy Syndromes
Axis 5: Impairment. Classification of impairment. Optional additional diagnostic parameter
Yet, a syndromic diagnosis is not always possible while presumed seizures types and syndromes might alter as new information becomes available. Furthermore, a classification system is used for a variety of purposes; epidemiological investigations, basic research, clinical aspects (e.g. screening patients before surgery) and clinical trials. Thus a classification system should be able to address several different needs of diverse areas. Therefore, in the light of new basic and clinical science advances and led by the need to further simplify terminology used, a new classification has arisen concerning mainly seizures classification but also introducing a simplification of terminology used on epilepsies (Tables 2.2 and 2.3) [26].
Table 2.2
Classification of seizures (2010)
Generalized seizures: Tonic-clonic; Absence (Typical, Atypical, Absence with special features); Myoclonic (Myoclonic, Myoclonic atonic, Myoclonic tonic); Clonic; Tonic; Atonic |
Focal seizures: according to severity (consciousness impairment, motor or autonomic components, subjective sensory or psychic phenomena, evolving to bilateral, convulsive seizures); according to site of origin according to sequence of clinical features |
Unknown |
Table 2.3
Electroclinical syndromes and other epilepsies (2010)
By age at onset |
Neonatal period (Benign familiar neonatal epilepsy; Early myoclonic encephalopathy; Ohtahara syndrome |
Infancy (Epilepsy of infancy with migrating focal seizures; West syndrome; Myoclonic epilepsy in infancy; Benign infantile epilepsy; Benign familiar infantile epilepsy; Dravet syndrome; Myoclonic encephalopathy in nonprogressive disorders |
Childhood (Febrile seizures plus; Panayiotopoulos syndrome; Epilepsy with myoclonic atonic seizures; Benign epilepsy with centrotemporal spikes; Autosomal-dominant nocturnal frontal lobe epilepsy; Late onset childhood occipital epilepsy; Epilepsy with myoclonic absences; Lennox-Gastaut syndrome; Epileptic encephalopathy with continuous spike and wave during sleep; Landau-Kleffner syndrome; Childhood absence epilepsy |
Adolescence-Adult (Juvenile absence epilepsy; Juvenile myoclonic epilepsy; Epilepsy with generalized tonic–clonic seizures alone; progressive myoclonus epilepsies; Autosomal dominant epilepsy with auditory features; other familial temporal lobe epilepsies |
Less specific age relationship (Familial focal epilepsy with variable foci; Reflex epilepsies) |
Distinctive (MTLE with HS; Rasmussen syndrome; Gelastic seizures with hypothalamic hamartoma; Hemiconvulsion-hemiplegia-epilepsy) |
Epilepsies not fitting into any of the above are separated by the presence or not of a known presumed cause (structural or metabolic) and by the primary mode of seizure onset (generalized or focal) |
By structural or metabolic causes |
(Malformations of cortical development; Neurocutaneous syndromes; Tumor; Infection; Trauma; Angioma; Perinatal insults; Stroke etc. |
Of unknown cause |
Conditions with epileptic seizures not diagnosed as epilepsy per se |
2.4 Etiology
The possible identification of specific brain abnormalities associated with epilepsy has crucial implications in the treatment and prognosis of the disease. A broad category of brain abnormalities stems from a strongly genetic/developmental component, while other cases mainly originate from acquired insults (such as infection, trauma and hypoxia). While still classified together with other acquired pathologies, hippocampal sclerosis is considered to originate from a combination of genetic risk factors and initiating insults (such as febrile seizures) [29].
Lesions such as tumors and severe trauma are in no way transient and reversible. Seizures occurring in patients with a potential cause of epilepsy (such as a highly epileptogenic oligodendroglioma) are not classified as acute symptomatic seizures [30]. Moreover, if the lesion generates an enduring predisposition for unprovoked seizures, with a risk comparable to those who have had two unprovoked seizures, then the person should be considered to have epilepsy despite having only one seizure [15].
Although there is an apparent variance in the power of potential seizure triggers, a recognized epilepsy syndrome comes to blur the lines. Seizures in reflex epilepsies are triggered by sensory, motor or cognitive stimuli, such as bright lights, eating and music. The pathophysiology of this syndrome involves the activation of hyperexcitable diffuse cortical pathways, with different triggering points according to the precipitating stimulus (f. ex. occipital triggering and propagation through cortico-cortical pathways in photosensitivity [31].
2.4.1 Febrile Seizures
Febrile seizures (FS) encountered in children are a well-known example of provoked seizures. Factors involved in their pathogenesis include genetic susceptibility (reflected in a positive family history), inflammatory mechanisms (with a particular significance of IL-1β) [32], mutations in the GABA-A receptors and participation of sodium channels [33]. FS are benign and not to be confused with the distinct epileptic syndrome of Generalizes Epilepsy with FS (GEFS+).
2.4.2 Developmental Brain Abnormalities
Abnormalities in neuronal cell migration and dysplasias of the neural ectoderm occur in embryonic life and result in malformations of cortical development (such as hemimesencephaly, heterotopias and lissencephaly) and epileptogenic neurocutaneous syndromes (Tuberous Sclerosis Complex and Sturge-Weber syndrome), respectively. A strong genetic component is involved in Tuberous Sclerosis (autosomal dominant inheritance) as well as in vascular malformations such as cavernomas. Such lesions mainly result in intractable seizures that require surgical intervention.
A variety of aggressors can disrupt the normal migration of neurons from the periventricular germinal matrix to their final destination and disrupt cortical lamination, thus resulting in a failure of normal circuit formation. These include infectious agents (such as rubella and the TORCH complex), toxins (e.g. alcohol) [34], while genetic factors may also play a role. Histologically, a scattering of large anomalous neurons is observed, associated with thickening and white matter “balloon cells” (focal cortical dysplasia), a blurring of the cortical/white matter border (hemimegalencephaly), while heterotopic gray matter islands in the white matter are observed in heterotopias. Lissencephaly and pachygyria are notable for disrupted gyri architecture [35].
In Tuberous Sclerosis, blurred cortical lamination results from masses of astrocytic cells and calcifications, while in Sturge-Weber cortical atrophy is the result of overlying angiomas. This finding is also present in vascular malformations. Other epileptogenic factors include the presence of hemosiderin from recurrent hemorrhages, especially in cavernomas [36].
2.4.3 Acquired Lesions
Epilepsies resulting from non-developmental abnormalities include post-traumatic epilepsy (PTE), seizures originating after hypoxic brain injury (as in stroke and perinatal insults) and tumor-associated epilepsy (TAE). Hippocampal sclerosis (HS) and medial temporal lobe epilepsy (MTLE), classified as a distinctive constellation, will also be mentioned here.
Although infectious and immunologic aetiologies are important in acquired epilepsy, with the former playing an important role in the developing world (mainly in the form of tuberculomas and cystic brain lesions due to neurocysticercosis) and the latter in surgical constellations such as Rasmussen’s Syndrome [35], a systematic review is beyond the scope of this chapter.
2.4.3.1 Brain Injury (Traumatic, Hypoxic/Ischemic)
Post traumatic epilepsy (PTE) is the most common cause of new-onset epilepsy in young adults [37] and accounts for 20 % of structural epilepsy (5 % of all epilepsy cases) [38]. Nevertheless, only a sub-group of brain trauma patients will develop epilepsy. The latency from the time of the injury to the onset of epilepsy is extremely variable (weeks to years) [39] while scalp EEG may be unable to detect initial epileptiform activity [40]. Higher injury severity and the presence of an intracranial hematoma are important risk factors for both early (in the first week after injury) and late seizures [41]. Other risk factors for PTE include advanced patient age, multiple concussions and seizures within 24 h post-injury [42].
Histopathological consequences of penetrating injuries include the formation of an epileptogenic cortical scar, while non-penetrating trauma results in axonal shearing, edema and ischemia in the gray–white matter junction [43]. Haemoglobin breakdown products, resulting from haemorrhage, have been implicated in epileptogenesis [44].
Changes in molecular signaling involving gene induction and modifications of neurotransmitter receptors and ion channels occur early after an injury. Axonal sprouting and dendritic modifications (such as mossy fiber sprouting) happen later on. A variety of mechanisms, including blood-brain barrier disturbances, inflammatory responses and release of related cytokines have also been implied [43]. All of the above changes result in an increased excitability that lowers seizure threshold.
Neuronal damage extends past an acute hypoxic insult and into the reperfusion phase. Pathophysiological mechanisms include apoptosis, activation of inflammatory mediators [45] as well as excessive extracellular glutamate excitotoxicity and intracellular accumulation of calcium [46]. Astrocytes post-insult release signals (such as thrombospondins) that increase excitatory synapse formation [47].
Apart from mechanisms leading to increased neuronal excitation, tissue necrosis and liquefaction can result in the formation of isolated cortical foci. Animal models of cortical isolation (“undercut” models) have shown a selective loss of GABAergic interneuron [48], resulting in a limitation of inhibitory mechanisms, which also contributes to epileptogenesis. It is worth noting that cortical isolation is present in both acquired (PTE, hypoxic injury) and developmental structural epilepsies.
2.4.3.2 Hippocampal Sclerosis (HS)
HS is present in at least 30 % of all epilepsy cases, according to both surgical [49] and post-mortem [50] series. HS is mainly associated with mesial temporal lobe epilepsy (MTLE) and reported in 50–70 % of patients with TLE and MTLE [51].
Whether HS is the cause or a consequence of seizures remains a highly controversial subject. Although status epilepticus has been proved to cause neuronal death, whether brief seizures amount to a similarly deleterious effect is still debated. Studies supporting the claim that the type of precipitating injury, seizure frequency and severity influence HS abound [52]. On the other hand, it has been proposed that hippocampal abnormalities in TLE play a pathogenetic role. Further support to this claim comes from imaging studies, showing that the degree of hippocampal atrophy is not correlated with the duration and severity of seizures [53]. Also, patients with multiple or poorly controlled seizures, as well as patients in a distinct subgroup of TLE, do not always display hippocampal sclerosis and neuronal loss. Finally, it has also been suggested that the pathologic alterations observed in sclerotic hippocampi can result from an abnormal cell migration during brain development [54].
Histopathologically, two areas of the hippocampus are mainly affected in HS: Ammon’s horn and the dentate gyrus. In Ammon’s horn, there is a marked neuronal depletion and astrocytic proliferation. These astrocytes display increased expression of glutamatergic receptors and are more capable of generating action potential-like responses in vitro compared to normal astrocytes [55]. In the dentate gyrus, loss and dispersion of granule cells, growth of new fiber systems (mossy fiber sprouting), loss of inhibitory interneurons and upregulation of inhibitory neurotransmission (perhaps in an attempt to curb increased excitation) are observed. In contrast to these changes, the subiculum (which is the main output region of the hippocampus) remains intact [56]. Nevertheless, it has been reported to contribute to epileptogenesis by initiating spontaneous interictal discharges [57].
ILAE classifies HS in three distinct categories, based on the regions (CA1 or CA4) involved in neural cell loss and gliosis. These categories differ in epilepsy history and prognosis of postsurgical seizure control [54].
2.4.3.3 Tumor-Associated Epilepsy (TAE)
Epilepsy is far more common in brain tumor patients than in the general population. Compared to high-grade tumors and cerebral metastases, low-grade gliomas are linked to a higher risk of seizures. Although the longer survival rate of patients with low-grade tumors, as well as molecular differences of each tumor category, could contribute to these findings, slow growing tumors might deafferentate cortical areas and thus create highly epileptogenic foci, as opposed to high-grade tumors that could induce epilepsy mainly through rapid tissue disruption and necrosis [58]. The most common seizure types in TAE are generalized tonic-clonic and complex partial seizures.
Evidence from both animal models [59, 60] and patients with TAE [61, 62] point towards the fact that crucial changes occur in the peritumoural region and not the immediate tumor invasion zone. These include glial cell swelling and damage resulting from hypoxia, acidosis [63] and altered expression of glutamate receptors in reactive astrocytes.
Numerous studies have shown that a frontal location of the tumor (and thus close to the premotor and motor cortex) is associated with a higher probability of associated epilepsy [64, 65]. Propagation of abnormal signals and/or a disruption of electrical activity transmitted to the primary motor cortex could be a logical explanation for the higher incidence of epilepsy when a tumor is located closer to the premotor cortex. In addition, the limbic and temporal lobe, the primary somatosensory cortex (S-I) and the opercula and insula regions of the secondary somatosensory area (S-II) also have a low threshold for producing seizures [65]. Notable exceptions include medial sphenoid wing meningiomas (where incidence of seizures is low) and seizure-like phenomena associated with posterior temporal-inferior occipital lesions [66].
2.4.4 Seizure Precipitants and Modulators
2.4.4.1 Sleep Deprivation
There is a controversy regarding seizures occurring after sleep deprivation. EEG recordings after sleep deprivation in epilepsy patients have been widely used, while sleep deprivation is known to activate epileptiform discharges independently of the activating effects of sleep [67]. However, seizures occurring after sleep deprivation are not classified as provoked by ILAE [15] and epilepsy specialists [68], since recurrence is far more likely than for patients with a provoked seizure. Further confounding to this issue, sleep deprivation is causing severe stress, a known seizure promoting factor, so it is hard to say whether epilepsy aggravation upon sleep deprivation is a direct effect (Chap. 3 will elaborate on the complex relationship between sleep and epilepsy).
2.4.4.2 Stress and Epilepsy
Seizures, particularly infantile spasms, have been linked to a stress-related elevation of CRH, which is responsible for the cascade leading to the production of adrenal steroids (such as cortisol). CRH has been found to act proconvulsively on seizure-prone brain regions such as the limbic system [69], by enhancing the actions of glutamate and suppressing afterhyperpolarisation [70]. Glucocorticoids limit the production of CRH as part of a negative feedback loop. Nevertheless, they activate CRH gene expression in the amygdala (a potential proconvulsive action) while adrenal-derived neuropeptides have both pro- and anticonvulsive properties [71]. Adrenocorticotropic hormone (ACTH) is released from the pituitary in response to hypothalamic CRH and has been proved to be anticonvulsive. ACTH also suppresses CRH, while inducing steroid synthesis, and thus is superior to oral steroids in the treatment of infantile spasms [72]. Under the influence of CRH, β-endorphin is released in an attempt of stress-level reduction. It acts as an endogenous opioid (μ-receptors), inhibiting the release of GABA and increasing dopamine levels [73] hence its excitatory effect.
2.4.4.3 Epilepsy and Reproductive Hormones
While men are reported to be more susceptible to epilepsy [74], sex-specific seizure propensity differs in various epilepsy syndromes. Sex-related differences are mainly thought to be the consequence of estrogen, progestin and androgens, which are also responsible for sexual brain dimorphism and behaviour patterns [75].
Ten to seventy percent of female patients with epilepsy has a greater propensity to seize in specific phases of their menstrual cycle, a “clustering” termed catamenial epilepsy, and observed in premenstrual and preovulatory periods, where the estrogen/progesterone ratio is high. Progesterone has antiepileptic properties, mediated by the GABAergic neurosteroid allopregnanolone, while it also has receptors in the limbic system. Estrogen, on the other hand, is primarily proconvulsive, by directly affecting glutamate receptor subtypes and increasing the density of hippocampal dendritic spines and excitatory synapses in animal models. Nevertheless, it has also been reported to increase neuropeptide Y concentration, with a possible antiepileptic effect [76].
2.5 Pathophysiology of Seizures and Epilepsy
Medicine is both art and science. The Hippocratic art of healing becomes a medical science to the extent that we succeed to identify which physiological function is changed in each disease and by what mechanism. Decades of neurological and neurosurgical observations have made numerous such links between the expression of seizures (loss of motor control, consciousness etc.) and the normal function of the particular brain areas which appear to initiate epileptic discharges and/or show lesions in each patient. In the process epileptology has become the greatest school for neuroscience, i.e. the description of the motor homunculus (for motor control), correlates of consciousness etc. More recently basic research in epileptology has elucidated many mechanisms underlying the expression of seizures at the macroscopic (brain imaging) and microscopic levels, especially in the ionic channels and neurotransmitter systems involved in the expression of seizures. Knowledge at the mesoscopic level (brain circuits) is seriously lagging behind. The most crucial question of mechanisms underlying epileptogenesis is still unanswered, although strides are currently made in this direction by molecular genetics and neuroplasticity research.
2.5.1 Basic Neurophysiology
GABA is the major inhibitory neurotransmitter in the brain. GABAA receptors coupled to chloride are involved to the hyperpolarizing of membranes which enables the inhibition of neuronal activity. The chloride mediated hyperpolarizing current counterbalances the excitatory input’s depolarizing currents and needs to be overcome for an action potential to fire. As data of animal models suggest, the acute GABAA receptor blockade will produce epileptic activity while in addition the GABAA receptors are the targets of many anticonvulsant medications which are currently in use. Benzodiazepines and topiramate increase the rate of chloride channels opening while barbiturates increase the duration that these channels remain open thus decreasing excitability. GABAB receptors show a similar action through a different mechanism. GABAB receptors couple to potassium channels and form a current with longer duration of action compared to the GABAA receptor activation [77].
Glutamate on the other hand, is a major excitatory neurotransmitter in the brain, mediated by three main receptors: N-methyl-d-aspartic acid (NMDA), alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA)/kainite and metabotropic. Selective NMDA receptor agonists show proconvulsant action while antagonists of the AMP/kainite and NMDA receptors have antiepileptic properties [78].
Glia cells and gap junctions between neuron cells facilitate voltage spread. GABA regulates the release and reuptake of neurotransmitters and transporters located both on glia and neurons. Furthermore, glia regulates the extracellular potassium concentration, which is correlated to neuronal excitability and epilepsy [79].
Two main types of ion channels exist. The ligand-gated channels which are activated by GABA, glutamate and acetylcholine, mediate cell communication at the synapse while the voltage-gated channels mediate the action potential and the axonal conduction of electrical signals. During resting state, the latter are closed and they open with the local membrane potential changes. Abnormalities of these channels lead to a disruption of membrane’s depolarization and repolarization that is necessary for the action potential. Several studies have suggested that channelopathies can be the underlying cause of certain epileptic syndromes. Generalized Epilepsy with Febrile Seizures Plus (GEFS+) is linked to missense mutations in sodium (NaV1.1) channels, while complete loss-of-function mutations in NaV1.1 cause Severe Myoclonic Epilepsy of Infancy. Benign Neonatal Infantile Seizures is also caused by mutations in sodium (NaV1.2) channels [80]. Potassium channelopathies have been implicated in Sudden Unexplained Death in Epilepsy (SUDEP, Kv1.1) [81], while monogenic idiopathic generalized epilepsies (IGE) have been associated with mutations in GABAA receptor genes [82]. Absence epilepsies have also been linked to GABAA [83], GABAB [84] as well as voltage-gated calcium channel dysfunction [85].
Neuromodulators (such as neuropeptides and neurosteroids) are endogenous factors that influence the balance between excitation and inhibition in the brain. Neuropeptides regulate GABAergic and glutamatergic neurotransmission, as well as the monoaminergic system. Neurosteroids are locally synthesized from cholesterol and circulating steroid hormones, and mostly target membrane receptors.
Many neuropeptides have been identified as endogenous antiepileptics. However, only one is currently in use as a treatment (ACTH). Other anticonvulsant neuropeptides include neuropeptide Y, which increases neocortical GABAergic neurotransmission and hippocampal dopamine, and somatostatin (expressed, most importantly, in GABA-ergic, inhibitory hippocampal hilar interneurons) [86].
The list of proconvulsant neuropeptides includes, among others, corticotropin-releasing hormone (CRH), β-endorphin and arginine-vasopressin peptide (AVP). While the former two are implicated in stress-related mechanisms, high levels of AVP have been associated with febrile seizures, given that this peptide in lower levels also acts as an endogenous antipyretic [87].
2.5.2 Hypersynchrony, Hyperexcitation and Epileptogenesis
Current research converges on the notion that seizures erupt when two fundamental measures of neuronal activity, excitability and synchronization, surpass certain normal level and that epileptogenesis is based on an aberrant exploitation of the most fundamental property of our brain, neuronal plasticity, the very one that enables us to develop, adopt to the environment and learn.
The early Penfield and Jasper’s hypothesis on generalized seizures introduced the idea of seizures correlating to synchronous brain activity characterized by decreased inhibition and enhanced excitation leading to the transient hypersynchronous activity of electrical epileptiform discharges [88]. The hallmarks of epilepsy which refer to hypersynchrony and hyperexcitation, still remain. Yet, synchronization in epilepsy is complex and it strongly depends on how ‘synchrony’ is defined, on the signals being measured (i.e. neuronal spikes or field potentials) and the spatial scale [89].
There is a variety of possible causes of hyperexitability. They can be intrinsic to neurons, like changes of types, numbers and opening times of voltage gated ion channels for Na+ or Ca2+, which may occur post-translationally, through 2nd messengers or through modulation of gene expression. They can also be extrinsic, like changes of local ion concentrations i.e. in [K+], and in extracellular space or modulation of transmitter metabolism or uptake by glial cells. Finally they can be synaptic, like increased excitatory post synaptic potentials (usually mediated by glutamate) and/or decreased inhibitory potentials (usually mediated by GABA), alterations in expression of transmembrane gated ionotropic channels, remodeling of synapse location or configuration, changes in gap-junction synaptic function etc. Simply put, it is a matter of balance tipped in favor of excitation in specific neurons (Fig. 2.1).
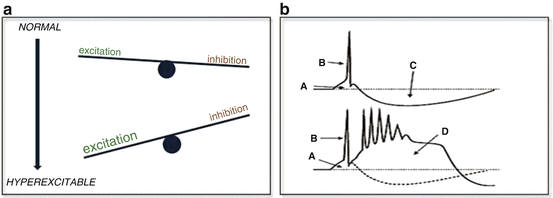
Fig. 2.1
 Source-Estimation from Non-invasive Recordings of Brain Electrical Activity in Sleep and Epilepsy
Source-Estimation from Non-invasive Recordings of Brain Electrical Activity in Sleep and Epilepsy
 Secure and Efficient WSN Communication Infrastructure
Secure and Efficient WSN Communication Infrastructure
 System Middleware
System Middleware
 Sleep Features and Underlying Mechanisms Related to Epilepsy and Its Long Term Monitoring
Sleep Features and Underlying Mechanisms Related to Epilepsy and Its Long Term Monitoring
 Offline Analysis Server and Offline Algorithms
Offline Analysis Server and Offline Algorithms
 DSMS and Online Algorithms
DSMS and Online Algorithms




(a) Simplified view of excitability changes towards an epileptiform activity. (b) Comparison of a normal neuronal discharge (upper) to an epileptiform one (lower). A = excitatory postsynaptic potential, B = sodium channel mediated action potential, C = inhibitory postsynaptic potential. Blockage of C leads to delayed and persistent expression of NMDA receptor channels mediated Ca++ spikes and paroxysmal depolarization shift (D)
Related posts:
Source-Estimation from Non-invasive Recordings of Brain Electrical Activity in Sleep and Epilepsy
Secure and Efficient WSN Communication Infrastructure
System Middleware
Sleep Features and Underlying Mechanisms Related to Epilepsy and Its Long Term Monitoring
Offline Analysis Server and Offline Algorithms
DSMS and Online Algorithms
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
