(1)
Cognitive Function Clinic, Walton Centre for Neurology and Neurosurgery, Liverpool, UK
6.1 Blood Tests
6.2 Neuroimaging
6.3 Neurogenetics
6.3.1 Alzheimer’s Disease
6.3.3 Other Genetic Disorders
6.4 Neurophysiology
Abstract
This chapter examines the utility of various investigative techniques in the diagnosis of cognitive disorders including, in order of increasing invasiveness, neuroimaging, neurogenetics, neurophysiology, cerebrospinal fluid analysis, and tissue diagnosis.
Keywords
DementiaDiagnosisInvestigationNeuroimagingNeurogeneticsNeurophysiologyCerebrospinal fluidThe investigation of cases of suspected dementia has perhaps gained a higher profile in recent years. This may reflect a combination of the increasing availability of more sophisticated investigation methods and the search for disease biomarkers which might be used as surrogates for pathological confirmation of disease. For example, recent diagnostic criteria for Alzheimer’s disease (AD; Dubois et al. 2007; Box 6.1; McKhann et al. 2011) enshrine investigation findings which examine potential AD biomarkers. Investigation findings are also integral to criteria for other dementing disorders, such as behavioural variant frontotemporal dementia (Rascovsky et al. 2011), Creutzfeldt-Jakob disease (Zerr et al. 2009), and corticobasal degeneration (Armstrong et al. 2013). Nevertheless, at time of writing dementia remains a clinical diagnosis. Diagnostic errors based on over-reliance on investigations, particularly structural imaging reported to show brain atrophy, have been encountered (Larner 2004a; Davies and Larner 2009).
Box 6.1: Proposed Diagnostic Criteria for Alzheimer’s Disease (After Dubois et al. 2007; Larner 2010)
Probable AD: Diagnosis requires A plus one or more supportive features B–E | |
Core criteria: | |
A | Early significant episodic memory impairment that includes: |
1. Gradual and progressive change in memory function over >6 months | |
2. Objective evidence of significantly impaired episodic memory on testing | |
3. Episodic memory impairment may be isolated or associated with other cognitive changes at AD onset | |
Supportive criteria: | |
B | Medial temporal lobe atrophy on MRI |
C | Abnormal CSF biomarker: ↓Aβ42, ↑total tau, ↑phospho-tau |
D | Specific pattern on functional neuroimaging with PET (NB not SPECT) |
E | Proven AD autosomal dominant mutation in the immediate family |
Definite AD: Requires clinical features + neuropathological confirmation, or clinical features + presence of deterministic genetic mutation | |
Despite caveats, it is reasonable to consider pursuing a number of investigations over and above “bedside” neuropsychological assessment (see Chap. 4) in cases of cognitive complaint and/or suspected dementia, based in part on guidelines issued by the American Academy of Neurology (AAN; Anonymous 1994; Knopman et al. 2001) and the European Federation of Neurological Societies (EFNS: Waldemar et al. 2007, Box 6.2; Hort et al. 2010a; Sorbi et al. 2012).
Evidence level | ||
|---|---|---|
Blood tests: “generally proposed as mandatory” | ESR, full blood count, electrolytes, calcium, glucose, renal and liver function tests, thyroid stimulating hormone | Good practice point |
Blood tests: “often required” | Vitamin B12, serology for syphilis, HIV, Borrelia | Good practice point |
Neuroimaging: structural | CT: to identify surgically treatable lesions and vascular disease | A |
Neuroimaging: structural | MRI: to increase diagnostic specificity | A |
Neuroimaging: functional | SPECT and PET: may be useful in those cases where diagnostic uncertainty remains | B |
Neurophysiology | EEG: useful adjunct, especially if CJD or transient epileptic amnesia suspected | B |
Cerebrospinal fluid | Cell count, protein, glucose, protein electrophoresis in atypical presentations | Good practice point |
Cerebrospinal fluid | Total tau, phospho-tau, Aβ42 as adjunct in cases of diagnostic doubt | B |
Genetic testing: known pathogenic mutations | In patients with appropriate phenotype or family history of autosomal dominant dementia. Only to be undertaken in specialist centres, with appropriate counselling of patient and family caregivers and with consent | Good practice point |
Genetic testing: ApoE | Not recommended as routine | B |
Tissue biopsy | For specific diagnosis of some rare dementias. Only to be undertaken in specialist centres | Good practice point |
6.1 Blood Tests
A variety of blood tests have sometimes been recommended as a “minimum dataset” for the investigation of suspected dementia cases. These include full/complete blood count, erythrocyte sedimentation rate, serum electrolytes, glucose, calcium, renal tests (urea, creatinine), liver-related blood tests (alkaline phosphatase, transaminases), red cell folate, serum vitamin B12, thyroid function and syphilis serology (Anonymous 1994; Bullock and Qizilbash 2002; Waldemar et al. 2007). In practice, many of these blood tests will already have been performed by primary care physicians prior to referral to the cognitive clinic as part of their “dementia screen”. If not, then it may be reasonable, following the AAN recommendations, to undertake measurement of vitamin B12 and thyroid function but reserve syphilitic serology for cases in which there are specific risk factors or a history of prior infection (Knopman et al. 2001). A counter argument has been made for syphilis screening in possible dementia cases in the developing world (Nitrini and Caixeta 2011). The EFNS guidelines of 2007 suggested that more extensive testing including vitamin B12 and serology for syphilis, HIV, and Borrelia might be required in individual cases (Waldemar et al. 2007). Arguments for (Nightingale et al. 2013) and against (Schott 2013) testing all dementia patients for HIV have been made.
A more recent EFNS guideline for AD seems to make vitamin B12 testing once again mandatory (Hort et al. 2010a), the various recommendations perhaps reflecting uncertainty about the diagnostic value of this test (its cost effectiveness is unproven: Marks 2011). Personally I am sceptical about vitamin B12 deficiency as a cause of cognitive impairment in anything other than very rare circumstances (Larner 2008a:194–5), with only one unequivocal case seen in nearly 20 years of neurological practice (Larner et al. 1999; Larner and Rakshi 2001). Low vitamin B12 levels may surely be an accompaniment of cognitive decline, perhaps related to poor dietary intake (weight loss is a common feature in early AD: Cronin-Stubbs et al. 1997), but without any anticipation of cognitive improvement with adequate repletion. This is, after all, often the situation with other complications of vitamin B12 deficiency, such as subacute combined degeneration of the spinal cord, peripheral and optic neuropathies (Larner 2002, 2004b; Larner et al. 1997). A trial of high dose vitamin B supplementation including vitamin B12 did not prevent decline in AD patients (Aisen et al. 2008).
The recommendations with respect to blood tests are prompted, at least in part, by the understandable desire to identify any potentially reversible cause(s) of cognitive decline, although in practice these are extremely rare (Clarfield 2003), with the possible exception of drug-related cognitive impairments, particularly related to the anticholinergic properties of some medications (Hejl et al. 2002).
Considering the large number of disorders which may result in cognitive impairment (Larner 2008a, 2013a), there are many other blood tests which may on occasion need to be considered, dependent on clinical context. From the perspective of potentially reversible causes, the autoimmune non-paraneoplastic limbic encephalitides, though rare, loom large. These may be associated with antibodies directed against LGI1 (previously known as voltage-gated potassium channel antibodies; Wong et al. 2010a), NMDA-receptors, and glutamic acid decarboxylase (GAD; Bonello et al. 2014).
Blood tests for genetic mutations causing inherited disorders of cognitive function are considered in Sect. 6.3.
6.2 Neuroimaging
Structural and functional neuroimaging modalities have been increasingly used in clinical practice to supplement clinical and cognitive assessment of patients with memory or other cognitive complaints. A wide array of imaging modalities is available (Jagust and D’Esposito 2009; Barkhof et al. 2011), although many of these are currently confined to the research arena. The original rationale for neuroimaging in this clinical situation was to exclude other possible, mostly structural, causes for cognitive decline (e.g. tumour, subdural haematoma, “normal pressure hydrocephalus”, dural arteriovenous fistula; white matter change) but the emphasis is now moving to the identification of markers of neurodegeneration in terms of the extent and location of brain atrophy and of pathological changes per se such as amyloid burden (e.g. PiB-PET, Klunk et al. 2004; florbetapir-PET, Clark et al. 2011).
6.2.1 Structural Neuroimaging: CT, MRI
Guidelines for the diagnosis of dementia have recommended the use of neuroimaging as a routine component of the initial evaluation of patients, as a supplement to clinical assessment in possible dementia cases (Knopman et al. 2001; Waldemar et al. 2007). The most recent EFNS guidelines recommend structural imaging in the evaluation of every patient affected by dementia (Filippi et al. 2012; Sorbi et al. 2012) whilst noting the difficulty of attributing clinical significance to evidence of cerebrovascular disease. Likewise national directives, such as the UK National Institute for Health and Clinical Excellence/Social Care Institute for Excellence (NICE/SCIE) guidelines (2006) state that “Structural imaging should be used in the assessment of people with suspected dementia to exclude other cerebral pathologies and to help establish the subtype diagnosis” (paragraph 1.4.3.2).
Various structural pathologies have been reported to cause dementia or cognitive impairment (Larner 2013a:166–76), including brain tumour (Case Studies 4.1 and 6.1; Ibrahim et al. 2009; Smithson and Larner 2013), subdural haematoma, and various causes of hydrocephalus, including colloid cyst (Case Study 6.2), but these have very rarely been encountered in CFC, accounting for less than 0.5 % of referrals (Larner 2013b).
Case Study 6.1: Clinical Utility of Structural Brain Imaging in Diagnosis of Dementia (1): Brain Tumour
A previously healthy 50 year-old man presented with a 4 month history of memory problems, word finding and naming difficulties, poor concentration, with associated anxiety and agitation. Neurological examination was normal. Neuropsychological assessment showed generalised intellectual loss (fullscale IQ decline of 35 points). The patient was impaired on all measures of auditory and visual memory for immediate and delayed recall, recognition memory and working memory. He made dysphasic errors, performed poorly on copy of the Rey-Osterrieth figure, and was severely impaired on verbal fluency and the Stroop test. Based on these results, a diffuse dementing illness was suspected, possibly Creutzfeldt-Jakob disease. Subsequent brain imaging disclosed a large left temporoparietal, non-homogeneous, space-occupying lesion with a large cystic component with surrounding oedema and mass effect. At surgery this proved to be an atypical meningioma grade II which was completely resected. Repeat neuropsychological assessment 4 months post-operatively showed significant improvement with no evidence of intellectual or memory impairment.
Case Study 6.2: Clinical Utility of Structural Brain Imaging in Diagnosis of Dementia (2): Colloid Cyst
A 65 year-old lady presented with her son who gave a 4 week history of declining memory and alertness in his mother such that his father had had to take over all the household duties. The patient was having difficulty walking, tending to shuffle, and had fallen on occasion. She was very sleepy during the day. A diagnosis of depression had been suspected in primary care and antidepressant medication started, without effect. Psychomotor retardation was evident on history taking and the head turning sign (Sect. 3.2.2) was present. In CFC a diagnosis of normal pressure hydrocephalus was mooted, but brain imaging (CT) showed a hyperdense lesion within the foramina of Monro representing a colloid cyst, with hydrocephalus and periventricular oedema, requiring prompt neurosurgical intervention.
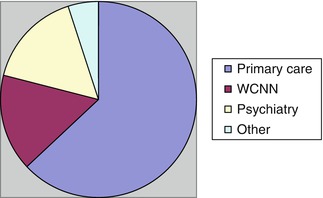
Because of the ready availability of structural neuroimaging, particularly computed tomography (CT), to primary care physicians as well as to cognitive neurologists, it might be argued this investigation should be undertaken prior to referral, as is the case for “screening blood tests”. A prospective observational study of 100 consecutive patients referred to CFC (March-August 2010) collected reports of neuroimaging performed prior to and after referral (Larner 2011a). Referral source (Fig. 6.1) was predominantly from primary care (63). Of the cases referred from secondary care (37), there were equal proportions from other neurology consultants at the Walton Centre for Neurology and Neurosurgery (WCNN; 16) and from psychiatry/old age psychiatry services (16), with a handful from other sources (5), such as general physicians.
The majority of patients were not demented by DSM-IV criteria (67), although there was evidence for a neurological or neurodegenerative process causing cognitive impairment in some of these non-demented individuals (mild cognitive impairment [MCI] 10, progressive nonfluent aphasia [PNFA] 2, dementia with Lewy bodies [DLB] 2, HIV 1). One third of patients received a clinical diagnosis of dementia (33), the most frequent subtypes being AD/Down syndrome (15), frontotemporal lobar degeneration syndromes (FTLD; 8), DLB (3), and vascular dementia (2).
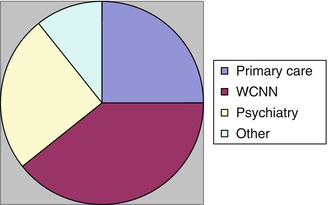
Patients who had undergone neuroimaging before referral to CFC were in the minority (28/100). Not unexpectedly, fewer primary care (7/63 = 11 %) than secondary care (21/37 = 57 %) referrals had undergone neuroimaging (Fig. 6.2), a statistically significant difference (χ2 = 26.0, df = 1, p < 0.01). In the group of referrals from secondary care, neuroimaging frequencies by referral source were: WCNN consultant 11/16 (=69 %); psychiatry/old age psychiatry 7/16 (=44 %); and others 3/5 (=60 %).
Breakdown of the frequency of neuroimaging by patient diagnosis showed that patients ultimately receiving a diagnosis of dementia from CFC were significantly more likely to have been imaged (15/33 = 45 %) than those not demented (13/67 = 19 %; χ2 = 8.14, df = 1, p < 0.01), although notably the latter group included some of those patients with underlying neurological and/or neurodegenerative disease (PNFA 2, HIV, DLB, MCI).
Following CFC consultation, all remaining patients (72) underwent neuroimaging. These data indicated that CFC was compliant with NICE/SCIE (2006) guidelines on neuroimaging. The key question though is whether this compliance makes any difference in clinical management. To a certain extent this may be a subjective value judgement, but it was the case that no revision of clinical diagnosis (e.g. not dementia revised to dementia, or a change of dementia subtype) was forthcoming as a result of CFC neuroimaging in this study (Larner 2011a), although this has happened on occasion (see Case Studies 4.1, 6.2, and 6.3). Finding potentially reversible causes of cognitive decline on neuroimaging is, sadly, exceedingly rare, the most common treatable structural causes seen in CFC being meningioma and intracranial dural arteriovenous fistula (three cases each in approximately 10 years; Wilson et al. 2010; Larner 2013b).
Case Study 6.3: Clinical Utility of Structural Brain Imaging in Diagnosis of Dementia (3): Diagnostic Revision
A 64 year-old lady presented with an 18-month history of forgetfulness, such as leaving the cooker on, with some consequent reduction in her household activities. Her primary care physician thought she might be depressed and prescribed an antidepressant. On the MMSE she scored 26/30 dropping single points for orientation in date, serial 7s, 5-min recall and intersecting pentagons. It was not clear whether these deficits related to depression or the early stages of a neurodegenerative disorder. Brain imaging with CT showed right frontotemporal atrophy, suggesting the possibility of FTLD. At reassessment, collateral history from her daughter indicated apathy and reduced personal hygiene. On the Frontal Assessment Battery (see Sect. 4.11) she scored 14/18 with impairments in lexical fluency, motor series, conflicting instructions, and go-no-go. MR brain imaging confirmed right frontotemporal atrophy and 1H-MR spectroscopy (Sect. 6.2.2) showed reduced N-acetyl aspartate and increased myoinositol in frontal but not occipital voxels. EEG was within normal limits. A diagnosis of behavioural variant frontotemporal dementia was therefore thought most likely. Neurogenetic testing for progranulin mutation (see Sect. 6.3.2) was negative.
A second prospective observational study of neuroimaging practice prior to CFC referral was undertaken on consecutive patients over a 6-month period (September 2012-February 2013). Of 127 patients (M:F = 70:57; 55 % male; age range 19–89 years, median 61 years), the majority were not demented by DSM-IV criteria (86, =68 %), giving a dementia prevalence of 32 %, but 32 had cognitive impairment short of dementia giving a prevalence of cognitive impairment of 57 %. As before, patients who had undergone neuroimaging before referral to CFC were in the minority (51/127 = 40 %), most initiated in secondary (46) care but a few (5) in primary care. However, the proportion imaged prior to referral had increased compared to the 2010 study (40 % vs 28 %). Breakdown of the frequency of neuroimaging by patient diagnosis showed that patients receiving a diagnosis of dementia were significantly more likely to have been imaged (23/41 = 56 %) than those not demented (28/86 = 33 %; χ2 = 7.38, df = 1, p < 0.01). Patients with cognitive impairment were significantly more likely to have been imaged (36/73 = 49 %) than cognitively healthy individuals (15/54 = 28 %; χ2 = 6.56, df = 1, p < 0.01).
So should neuroimaging be a prerequisite for referral to memory clinics, part of the minimum dataset available to the CFC clinician prior to assessment? Probably not.
A steady trickle of patients is referred to CFC because of “cortical atrophy”, based on radiology reports of brain scans undertaken for other purposes, usually headache. (Twas ever thus: Allison (1962:257) reports a patient diagnosed with early AD because of symmetrical ventricular dilatation on air encephalography, an investigation now obsolete.) Although this label of “atrophy” may be technically correct according to neuroradiological terminology (Global Cortical Atrophy scale, grade 1 = opening of sulci), it is invariably a qualitative judgement. Moreover, experience indicates that it is seldom of clinical relevance, and serves only to generate significant (and understandable) anxiety in both patients and primary and secondary care physicians unfamiliar with the uses of neuroimaging in the assessment of cognitive problems. As with all imaging findings, clinical-radiological correlation is essential, which basically privileges the primacy of clinical assessment. A study of the diagnostic yield of CT scans done routinely in a UK memory loss clinic found “significant findings” in only 1 % (Dawe 2012). Neuroimaging provided support for, but did not alter, clinical diagnosis in a large clinico-pathological case series (Snowden et al. 2011).
With magnetic resonance imaging (MRI), the problem of incidental findings may be even more evident since they are very common (Morris et al. 2009). The presence of cerebrovascular disease in particular is problematic, as acknowledged in EFNS guidelines (Sorbi et al. 2012). Such changes seem to be interpreted by psychiatrists and old age psychiatrists as commensurate with a diagnosis of vascular dementia, despite the clarity of diagnostic criteria which require such changes to be clinically and temporally relevant to cognitive decline (Román et al. 1993; van Straaten et al. 2003; Gorelick et al. 2011).
Another incidental finding is brain calcification, particularly of the basal ganglia, said to occur in perhaps 0.5–1 % of normal CT scans, and in association with a variety of disorders (Larner et al. 2011:56–7), including Down’s syndrome (Case Study 6.4).
Case Study 6.4: Clinical Utility of Structural Brain Imaging in Diagnosis of Dementia (4): Diagnostic Confusion from Incidental Findings
A 48 year-old lady with Down’s syndrome presented to intellectual disability services with a 3-year history of decline in her abilities as reported by her carers. Brain imaging (CT) was undertaken, and was reported by a general radiologist to show Fahr’s disease, prompting urgent referral to CFC. The scan showed extensive symmetrical calcification of the basal ganglia, a striking example of a phenomenon well-described in Down’s syndrome (Takashima and Becker 1985; Mann 1988), without the need to invoke a diagnosis of Fahr’s disease. Indices of calcium metabolism were within normal limits.
Focal atrophy of the medial temporal lobe (MTA) on MRI is one of the supportive criteria in recent diagnostic criteria for AD (e.g. Dubois et al. 2007; Box 6.1). MTA discriminates pathologically confirmed AD from DLB and vascular cognitive impairment (Burton et al. 2009). Visual assessment scales for MTA are available (Barkhof et al. 2011:22–3) although such assessment is not routinely undertaken at present in this centre. Structural neuroimaging is often unrewarding in suspected cases of FTLD, particularly early in the disease course. In a benign and good prognosis variant of behavioural variant frontotemporal dementia (bvFTD), or bvFTD phenocopy, neuroimaging may change little over time (Davies et al. 2006).
Other MR based modalities, such as diffusion weighted imaging (DWI), susceptibility weighted imaging (SWI), and diffusion tensor imaging (DTI) may possibly be of value in the assessment of cognitive disorders (Barkhof et al. 2011). DWI may be of particular value in suspected cases of sporadic Creutzfeldt-Jakob disease (e.g. Ali et al. 2013), SWI in cerebral amyloid angiopathy (Charidimou et al. 2012), and DTI in white matter disorders (Filley 2012). Longitudinal volumetric MR imaging measuring rates of whole-brain and hippocampal atrophy has proved a sensitive marker of neurodegeneration and may well find increasing use as a diagnostic tool and as a surrogate marker to assess treatment effects (Frisoni et al. 2010).
6.2.2 Functional Neuroimaging: HMPAO-SPECT, 1H-MRS
Of the various functional imaging modalities, 99mTechnetium hexamethylpropylene amine oxime single photon emission computed tomography (99mTc HMPAO-SPECT) is probably the most widely available, although modern diagnostic criteria for AD specify functional brain imaging with positron emission tomography (PET) in preference to SPECT (Dubois et al. 2007; Box 6.1). In vivo biomarkers for diagnostic imaging may become of greater relevance in the future, such as fluorinated PET ligands for imaging amyloid (Klunk et al. 2004; Clark et al. 2011). There is no experience of PET in CFC.
Use of HMPAO-SPECT in the assessment of cognitive problems has been standard practice in some centres, usually based on visual analysis of regional cerebral blood flow changes. Although more sophisticated analytical techniques are possible, such as statistical parametric mapping, these are not universally available. Hypoperfusion of posterior temporal and parietal regions with relative sparing of occipital blood flow is typical of AD as compared to normal controls (e.g. Montaldi et al. 1990; Talbot et al. 1998; Dougall et al. 2003), although in the visual variant of AD (posterior cortical atrophy) occipital hypoperfusion is seen. Otherwise, occipital hypoperfusion is more typical of dementia with Lewy bodies (e.g. Lobotesis et al. 2001). HMPAO-SPECT additionally has the facility for the differential diagnosis of AD and FTLD based on the frontal hypoperfusion in the latter (e.g. Talbot et al. 1998; Charpentier et al. 2000).
The most recent EFNS guidelines suggested that SPECT perfusion imaging is useful to distinguish DLB, corticobasal syndrome and Creutzfledt-Jakob disease from AD (Sorbi et al. 2012:1173).
There has been only limited experience of functional neuroimaging in CFC, perhaps surprisingly in light of the origins of isotope imaging in Liverpool (Ansell and Rotblat 1948). This has been for both logistic (no SPECT scanner on site, nearest facilities at Royal Liverpool University Hospital and Wrexham Maelor Hospital) and financial reasons. A study of the utility of HMPAO-SPECT was performed in a cohort of young cognitively-impaired patients in whom diagnostic uncertainty remained after standard clinical and neuropsychological assessment and structural brain imaging (Doran et al. 2005a). SPECT scans were visually assessed by five raters (two consultant neurologists with a specialist interest in cognitive disorders, three nuclear medicine specialists) on two occasions 6 months apart, firstly without any clinical data (“blind”), secondly with brief pertinent clinical information (“informed”). SPECT diagnoses were compared with criterion diagnoses subsequently established by the two neurologists with access to all the clinical, neuropsychological and neuroimaging data. Despite reasonable intra- and inter-rater reliability, diagnostic accuracy ranged from 32 to 58 %. SPECT scan normality or abnormality in blind and informed viewings gave respective sensitivities of 77 and 71 %, specificities of 44 and 38 %, positive predictive values of 88 and 87 %, and negative predictive values of 27 and 18 %. Calculating pairwise disease group comparisons, likelihood ratios suggested some diagnostic gain in differentiating AD from “not AD” (as also shown by Dougall et al. 2003), and in differentiating AD from FTD/focal syndromes (as also shown by Talbot et al. 1998; McNeil et al. 2007). SPECT scanning was of little help in establishing diagnoses in this (highly selected) cohort of patients, a finding which supported the conclusion of an AAN evidence-based review of SPECT imaging which concluded that SPECT could not be recommended for either the initial or the differential diagnosis of suspected dementia because it had not demonstrated superiority to clinical criteria (Knopman et al. 2001). That said, other studies in less selected cohorts than that examined here have reached different, more positive, conclusions (Talbot et al. 1998; Salmon et al. 2009).
Dopaminergic SPECT imaging (FP-CIT, DATScan), for visualisation of the dopamine transporter, may be useful to differentiate AD from DLB (Hort et al. 2010a; Sorbi et al. 2012). However, it does not pick up all DLB cases (Colloby et al. 2012). Again, logistic and financial reasons have meant that this modality has been infrequently used in CFC patients with cognitive impairment (e.g. Ali et al. 2010).
Proton magnetic resonance spectroscopy (1H-MRS) is a form of functional neuroimaging which been advocated as a possible tool for use in the evaluation and diagnosis of dementia (Kantarci 2007), based on changes in the neuronal marker N-acetyl aspartate (NAA) and in myoinositol (mI) relative to brain creatine (Cr). NAA:Cr ratios are typically decreased in disorders characterised by neuronal loss, such as AD and FTLD, and mI:Cr ratios are elevated in the presence of pathological gliosis, which may also be seen in AD and FTLD.
The utility of 1H-MRS in the diagnosis of dementia has been assessed in a highly selected population of patients attending CFC (Larner 2006). Single voxel 1H-MRS was performed on GE Signa 1.5 T Scanner (TE = 35 ms; TR = 1,500 ms), measuring NAA and mI with Cr as reference, hence generating NAA:Cr and mI:Cr ratios. Comparison of mean NAA:Cr and mI:Cr ratios in occipital and frontal voxels in demented (n = 11; AD 8, FTLD 3) and non-demented (9) patients was performed. There was a statistically significant increase in mI:Cr ratio in occipital voxels in demented patients (t = 4.60, df = 15, p < 0.001), and a trend towards reduced NAA:Cr ratio in frontal voxels in demented patients (t = 1.86, df = 13, 0.05 < p < 0.1). Acknowledging the small patient numbers and the clinical heterogeneity of cases, nonetheless this study suggested that a high occipital mI:Cr ratio may be useful in differentiating demented from nondemented patients, and a low frontal NAA:Cr may be suggestive of a diagnosis of dementia.
A subsequent study assessed the utility of 1H-MRS in the differential diagnosis of AD (n = 9) and FTLD (n = 6; bvFTD 4, semantic dementia 2) (Larner 2008b). Occipital NAA:Cr ratio was lower in AD than FTLD patients, the difference reaching statistical significance (t = 2.47, df = 13, p < 0.05), but occipital mI:Cr ratio showed no difference between the groups (t = 0.81, df = 12, 0.1 < p < 0.5). Reduced occipital NAA:Cr ratio may reflect occipital neuronal loss occurring in AD, but not in FTLD, whilst the failure of occipital mI:Cr ratio to differentiate the two conditions may reflect either the absence of occipital gliotic change in FTLD or the equality of such change in both AD and FTLD (Larner 2008b). These latter findings contrast with the previously reported utility of mI:Cr ratio in differentiating cases of dementia (increased) from non-dementia patients (Larner 2006).
In a single case, extremely high frontal mI:Cr ratio, suggesting profound gliosis, prompted a clinical diagnosis of progressive subcortical gliosis of Neumann (Neumann and Cohn 1967), in which prior structural neuroimaging had suggested a provisional diagnosis of vascular change (subcortical arteriosclerotic encephalopathy) (Larner et al. 2003). Neuroaxonal leukodystrophy also entered the differential diagnosis of this case. Case Study 6.3 provides a further example of the diagnostic utility of 1H-MRS.
6.3 Neurogenetics
There has been a huge expansion in the understanding of the genetic causes of dementia in the past two decades (Box 6.3). This has had an increasing impact on clinical practice, admittedly in selected cases (Adab and Larner 2006; Aji et al. 2013a, b; Doran and Larner 2004a, b, 2006, 2009; Doran et al. 2007; Larner 2003, 2004c, 2007a, 2008c, d, e, 2009a, b, c, 2011b, c, 2012a, b, c, 2013c, d; Larner and Doran 2006a, b, 2009a, b, c; Larner and du Plessis 2003; Larner et al. 2007; Sells and Larner 2011; Ziso et al. 2014a, b).
Box 6.3: Some Monogenic Mendelian Causes of Dementia with Deterministic Genes and Online Mendelian Inheritance in Man (OMIM) Numbers (See Also Larner 2013a:110–44)
Familial Alzheimer’s disease:
Amyloid precursor protein (APP): OMIM#104300
Presenilin 1 (PSEN1): OMIM#607822
Presenilin 2 (PSEN2): OMIM#606889
Frontotemporal lobar degenerations:
Microtubule associated protein tau (MAPT): OMIM#600274
Progranulin (GRN): OMIM#607485
Valosin-containing protein (VCP): OMIM#167320
Charged multivesicular body protein 2B (CHMP2B): OMIM#600795
TAR-DNA binding protein 43 (TDP-43): OMIM#612069
Fused in sarcoma (FUS): OMIM#608030
C9ORF72: OMIM#105550
Familial prion diseases: all due to prion protein gene (PRNP) mutations:
Familial Creutzfeldt-Jakob disease: OMIM#123400
Gerstmann-Straussler-Scheinker disease: OMIM#137440
Fatal familial insomnia: OMIM#600072
Huntington’s disease-like 1: OMIM#603218
Huntington’s disease: IT15 gene, OMIM#143100
CADASIL: Notch 3 gene, OMIM#125310
In certain circumstances, neurogenetic testing may be indicated following appropriate genetic counselling, the exact nature of which is dependent upon whether testing is diagnostic (i.e. in a symptomatic individual) or predictive (i.e. in an asymptomatic individual with a suggestive family history). The latter may be best administered through the auspices of a clinical genetics service (Larner 2007b), perhaps using the model which is already well developed for families with Huntington’s disease. All studies of the genetic basis of dementia should observe appropriate consensus statements and guidelines emanating from national and professional bodies regarding genetic counselling and informed consent (Olde Rikkert et al. 2008; Goldman et al. 2011). Due to allelic heterogeneity, genotype-phenotype correlations are in their infancy for a number of these genes (Larner 2004c, 2013c; Larner and Doran 2006a, 2009a, b). Few examples of dementia resulting from genetic (monogenic Mendelian) mutations have been encountered in CFC (Doran and Larner 2009).
Routine use of genetic tests is not recommended (Knopman et al. 2001), testing or screening being reserved for families with the appropriate phenotype or with a family history of dementia transmitted in an autosomal dominant pattern (Hort et al. 2010a; Sorbi et al. 2012). The latter may be defined as at least three affected individuals in at least two generations (Cruts et al. 1998). However, experience with eight families with either PSEN1 or MAPT mutations seen in CFC (see below) showed that in only three was there a clear autosomal dominant pattern of disease transmission according to this definition; in four other families there was familial disease (i.e. at least one first degree family relative affected; Cruts et al. 1998); and one case was apparently sporadic, possibly due to de novo mutation (Larner and du Plessis 2003). All but one of these families had early onset dementia (before 65 years of age) (Doran and Larner 2009).
6.3.1 Alzheimer’s Disease
Mutations in three genes have been reported to be deterministic for AD (Box 6.3): amyloid precursor protein (APP), presenilin 1 (PSEN1), and presenilin 2 (PSEN2). Of these, the most commonly affected is PSEN1 (Alzheimer Disease and Frontotemporal Dementia Mutation Database 2014). PSEN1 mutations are associated with a variable clinical phenotype which may encompass not only cognitive decline but also epileptic seizures, myoclonus, extrapyramidal features, spastic paraparesis, behavioural and psychiatric symptoms sometimes akin to those seen in bvFTD, aphasia, agnosia, and cerebellar ataxia (Larner 2011b, 2013c; Larner and Doran 2006a, b, 2009b).
Four families with PSEN1 AD have been seen in CFC, with the following mutations: p.Tyr115Cys (Doran and Larner 2006), p.Met139Val (apparently a de novo mutation; Larner and du Plessis 2003), p.Arg269Gly (with prominent behavioural and psychiatric symptoms; Doran and Larner 2004b); and p.Arg269His (with late age at onset; Fig. 6.3; Larner et al. 2007). It is recognised that there may be underascertainment of such families in the UK (Stevens et al. 2011). Concurrence of early- and late-onset AD in some families (Brickell et al. 2006) may indicate a PSEN1 mutation (Lladó et al. 2010).
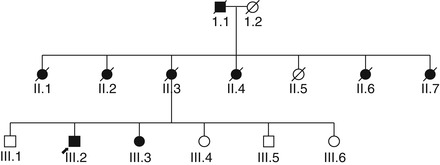
Families with late-onset AD apparently transmitted as an autosomal dominant condition but without any of the known AD mutations have been reported on occasion (e.g. Jimenez-Escrig et al. 2005). One such family has been encountered in CFC, with seven affected individuals in two generations, all with onset after age 65 years as far as could be ascertained from oral family history (Hancock and Larner 2007).
AD phenotype has also been reported with some tau (MAPT) gene mutations (Sect. 6.3.2) including p.Arg406Trp (Rademakers et al. 2003; Tolboom et al. 2010) and the IVS10+16C>T splice site mutation (Doran et al. 2007).
Related posts:
 History and Neurological Examination
History and Neurological Examination
 Management
Management
 Diagnosis (1): Cognitive Syndromes, Comorbidities, and No Diagnosis
Diagnosis (1): Cognitive Syndromes, Comorbidities, and No Diagnosis
 Assessment with Non-cognitive Screening Instruments and Combinations of Scales
Assessment with Non-cognitive Screening Instruments and Combinations of Scales
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


