Chapter 18 Involuntary Movement Disorders
The Basal Ganglia
Five subcortical gray-matter macroscopic nuclei (Figs 18-1 and 18-2) constitute the basal ganglia:
• The caudate nucleus and putamen, which together constitute the striatum
• The globus pallidus, which, together with the putamen, constitutes the lenticular nuclei
• The subthalamic nucleus (corpus Luysii)
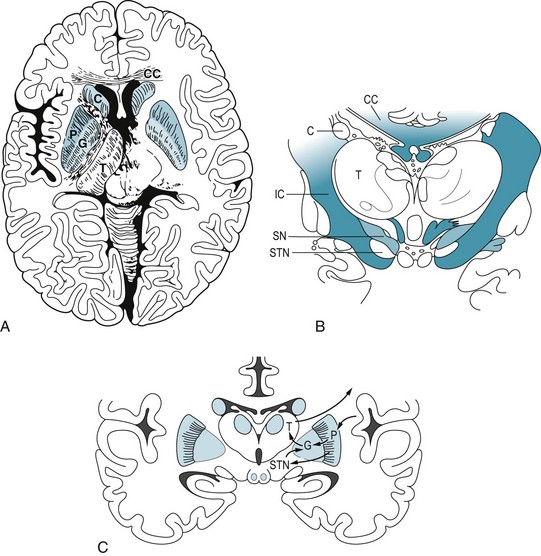
FIGURE 18-1 A, This axial view, the one used in computed tomography and magnetic resonance imaging studies, shows the basal ganglia in relation to other brain structures. The heads of the caudate nuclei (C) indent the lateral undersurface of the anterior horns of the lateral ventricles. The caudate and putamen (P) constitute the striatum. The globus pallidus (G), which has internal and external segments, and the putamen form the lenticular nucleus, named for its resemblance to an old-fashioned lens (see Fig. 18-1, C). The posterior limb of the internal capsule (IC) separates the lenticular nucleus from the thalamus (T), which is not a member of the basal ganglia family. B, In this coronal view of the diencephalon, the substantia nigra (SN), as well as the subthalamic nuclei (STN), sits below the thalamus. The substantia nigra, because of its characteristic shape and pigmentation, serves as a landmark. The lateral ventricles are bounded laterally by the heads of the caudate nuclei (C) and superiorly by the corpus callosum (CC). This myelin stain has blackened the heavily myelinated fibers of the internal capsule (IC) and corpus callosum (CC). C, A coronal view shows extrapyramidal circuits. The putamen sends a direct and an indirect dopamine tract to the internal segment of the globus pallidus (GPi). Dopaminergic neurons in the substantia nigra project to the putamen, where neurons with D1 receptors project directly to the GPi. Putaminal neurons with D2 receptors project through the globus pallidus external segment (GPe) and subthalamic nucleus and thence to GPi. The GPi innervates the ventrolateral nucleus of the thalamus, which projects to the motor cortex. The cortex, completing a circuit, innervates the putamen.
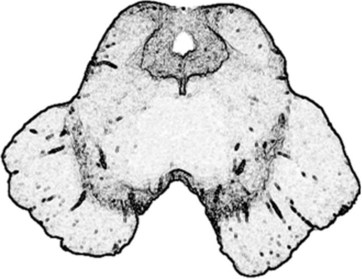
FIGURE 18-2 This computer-generated rendition of the midbrain should be compared to a photograph (see Fig. 2-9), functional drawings (see Figs 4-5 and 4-9), and an idealized sketch (see Fig. 21-1). The lower third of the midbrain, which lies just caudal to the diencephalon, contains the pair of horizontal but gently curved, elongated, pigmented nuclei – the substantia nigra. In Parkinson disease, the substantia nigra and other pigmented nuclei lose their pigment and, compared to normal, thus appear blanched. The midbrain also houses the prominent aqueduct of Sylvius, which is the dorsal heart-shaped hole surrounded by the periaqueductal gray matter. Cerebrospinal fluid passes from the third ventricle, through the aqueduct, to the fourth ventricle.
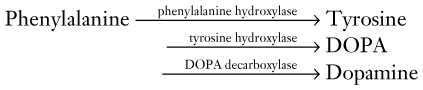
Although the extrapyramidal tract seems to play merely a supportive role – indirectly influencing the corticospinal tract by acting on thalamocortical connections and projecting only within the brain – it comprises several clinically important tracts. The most important one, from a clinical viewpoint, is the nigrostriatal tract, which, as its name suggests, extends from the substantia nigra to the striatum (Fig. 18-3). This tract provides dopamine innervation directly and indirectly, via the subthalamic nucleus, to the globus pallidus’ internal segment (GPi).
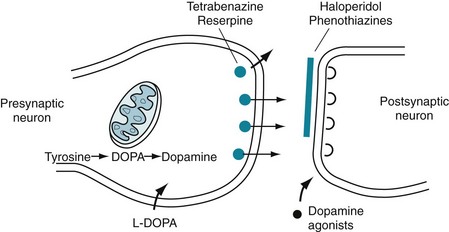
FIGURE 18-3 A succession of enzymes normally converts tyrosine to dopamine in the presynaptic nigrostriatal neuron. In Parkinson disease, the nigrostriatal tract degenerates, but the postsynaptic dopamine receptors remain intact. As a result of the degeneration, an absence of tyrosine hydroxylase leads to insufficient dopa and then markedly reduced synthesis of dopamine. L-dopa, which is a standard oral medication for Parkinson disease, penetrates the blood–brain barrier and substitutes for the deficient endogenous dopa in dopamine synthesis. D-dopa, the dextro-isomer, does not cross the blood–brain barrier and cannot enter the synthetic pathway. It is therefore useless as a treatment for Parkinson disease.
One of the main differences between dopamine receptors is that, in the striatum, dopamine binding to dopamine 1 (D1) receptors stimulates adenyl cyclase activity, but dopamine binding to dopamine 2 (D2) receptors inhibits adenyl cyclase activity (see Table 21-1).
General Considerations
The involuntary movement disorders share several clinical features. Anxiety, exertion, fatigue, and stimulants (including caffeine) increase the movements, but willful concentration and sometimes biofeedback may suppress them, at least transiently. Most movements disappear during sleep. The exceptions – hemifacial spasm, myoclonus, palatal tremor, tics, and specific sleep-related disorders, such as restless legs syndrome (RLS) and periodic movements – persist without regard to sleep stage (see Chapter 17).
Neurologists typically measure four parameters of involuntary movements:
• Hypokinesia or hyperkinesia. (For most clinical purposes, Parkinson disease and parkinsonism [see later] constitute the sole hypokinetic involuntary movement disorder.)
• Continuous or intermittent, discrete movements
• Generalized or focal involvement
Another error may occur when patients, at first glance, appear to have a psychogenic movement disorder (see later and Chapter 3). In many situations, the lack of a definitive confirmatory laboratory test forces neurologists to rely exclusively on their clinical judgment.
Parkinson Disease
The initial and ultimately most disabling physical feature of Parkinson disease is usually bradykinesia or, in the extreme, akinesia. Slow or absent movement produces the classic masked face (Fig. 18-4), paucity of trunk and limb movement (Figs 18-5 and 18-6), and impairment of activities of daily living. Parkinson patients sometimes liken their slow movements to slogging through hip-deep mud, wearing lead clothing, or driving a car with an engaged emergency brake.

FIGURE 18-4 Compared to normal individuals of the same age, Parkinson disease patients blink less frequently, offer fewer facial expressions, and less often move their head. Neurologists have called patients’ facial appearance a “stare” or “masked facies” (Latin, face or countenance), but now they usually describe the face as masked. Even when subtle, the masked face gives the appearance of apathy or depression.
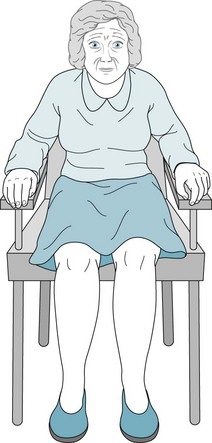
FIGURE 18-5 Parkinson disease patients typically sit motionless with their legs uncrossed and their feet flat. Their arms remain on the chair or in their lap and rarely participate in normal gestures or repositioning movements. In contrast to normal individuals and especially those with chorea, Parkinson disease patients do not shift their weight from one hip to another or make any unnecessary movements.
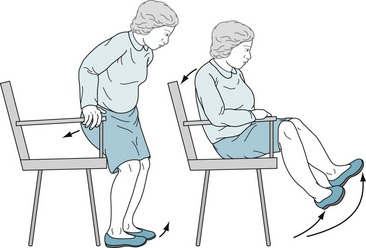
FIGURE 18-6 Patients with akinesia and rigidity cannot rapidly flex their spine, hips, or knees. When sitting, they tend to fall slowly and solidly backward into a chair. Unable to bend rapidly, their feet rise several inches off the floor. Sitting and turning en bloc signal early parkinsonism.
Rigidity typically accompanies bradykinesia (Fig. 18-7). Although rigidity is one of the cardinal features of Parkinson disease, it often appears as a manifestation of other extrapyramidal disorders. No matter the context, physicians should not confuse rigidity with spasticity, which signals corticospinal tract disease (see Chapter 2).
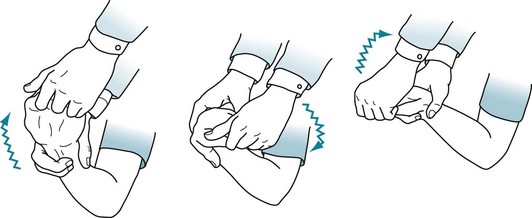
FIGURE 18-7 Physicians typically elicit rigidity by rotating the patient’s wrist. When present, rigidity causes an increased tone in all directions of movement. A superimposed tremor adds a ratchet-like, cogwheel rigidity resistance.
Tremor is often the most conspicuous feature of Parkinson disease; however, it is the least specific sign, least debilitating symptom, and least associated with dementia and depression. In Parkinson disease, the affected body part usually oscillates in a single plane with a regular rate, although with a variable amplitude. It primarily involves the hands. Even more characteristically, the tremor appears when patients rest quietly with their arms supported. Neurologists call it a resting tremor (Fig. 18-8) and can show that it differs from cerebellar and essential tremors. When patients have tremor as their primary symptom, neurologists refer to them as having “tremor-predominant” Parkinson disease.
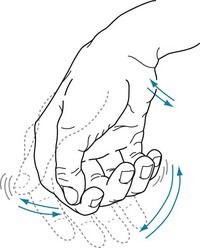
FIGURE 18-8 The resting tremor – a cardinal feature of Parkinson disease – consists of a relatively slow (4–6-Hz) to-and-fro flexion movement of the wrist, hand, thumb, and fingers most apparent when patients sit comfortably. Its similarity to shaking pills in a cupped hand gave rise to the description “pill-rolling” tremor. The tremor is exaggerated or sometimes apparent only when patients are anxious. However, voluntary movement or intense concentration may momentarily reduce the tremor, and sleep abolishes it. Rigidity and akinesia almost always accompany the Parkinson disease resting tremor.
Additional symptoms and signs may emerge as Parkinson disease advances. Patients lose their postural reflexes, which are neurologic compensatory mechanisms that adjust muscle tone in response to change in position. Loss of these reflexes, in combination with akinesia and rigidity, results in a characteristic gait impairment, marche à petit pas or festinating gait, which consists of a tendency to lean forward and accelerate the pace (Fig. 18-9, A). In a test of postural reflexes, the pull test, the examiner pulls the patient from the shoulders (Fig. 18-9, B). Normal individuals merely sway. Patients who have mild impairment of their postural reflexes take a few steps back, i.e., have retropulsion. More severely affected ones rock stiffly backwards without flexion or other compensatory movement and topple, en bloc, into the examiner’s arms.
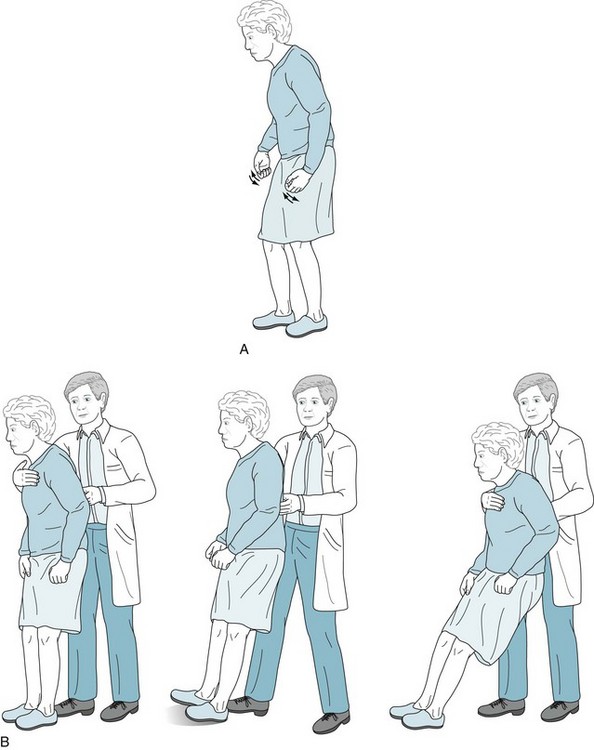
FIGURE 18-9 A, Parkinson disease often produces a festinating gait, in which patients take short, shuffling steps and accelerate their pace. Their neck and lower spine are typically flexed. When walking, they fail to swing their arms, look about, or have other normal accessory movements. Likewise, while turning en bloc, they simultaneously move their head, trunk, and legs. B, The pull test consists of the physician gently but rapidly pulling the patient’s shoulders. Unaffected individuals will compensate by taking one or two steps backward. Parkinson patients, who generally have impaired postural reflexes, will take many steps backward (i.e., have retropulsion), because they are unable to stop through reflexly weight-shifting compensatory maneuvers. In pronounced cases, as the one pictured here, patients unable to alter their posture will tilt backwards en bloc and fall into the physician’s arms. (Progressive supranuclear palsy patients have similar bradykinesia, lack of accessory movements, and a positive pull test, but their posture is extended or erect rather than flexed [see Fig. 7-7].)
Even at the onset of the illness, patients’ handwriting deteriorates to a small and tremulous script, micrographia (Fig. 18-10). In a parallel fashion, their voice loses both volume and normal fluctuations in pitch and cadence, i.e., their speech becomes hypophonic and monotonous. Also, because of their illness or the medications used to treat it, Parkinson disease patients develop sleep disturbances, characteristically rapid eye movement sleep behavior disorder (see Chapter 17).

FIGURE 18-10 The handwriting in this sample from a Parkinson disease patient shows progressive decrease in height (micrographia) and a superimposed tremor. These abnormalities in signatures, such as those on checks, can often date the onset of Parkinson disease. Although essential tremor may also cause tremor in handwriting samples, micrographia indicates that the underlying illness is Parkinson disease.
Nonmotor physical problems also emerge. For example, Parkinson patients characteristically lose their sense of smell. The anosmia, which also occurs in Alzheimer disease, reflects the neurodegenerative nature of these illnesses (see Chapters 4 and 7). They also routinely develop problems with their autonomic nervous system, including dysphagia, constipation, urinary incontinence, and abnormal sweating.
Psychiatric Conditions Comorbid with Parkinson Disease
Treatment of Comorbid Depression
Although SSRIs carry fewer side effects than TCAs in the population of depressed Parkinson disease patients, they may cause a unique problem. Prescribing SSRIs in conjunction with selegiline (deprenyl [Eldepryl]) can theoretically cause the serotonin syndrome because SSRIs prevent serotonin reuptake while selegiline, a monoamine oxidase (MAO) inhibitor, prevents its breakdown (see Chapter 6). The actual incidence of serotonin syndrome is very low because selegiline, in the doses used for Parkinson disease treatment, selectively inhibits only MAO-B, which metabolizes dopamine; whereas the serotonin syndrome is mostly a complication of inhibition of MAO-A, which metabolizes serotonin. Similarly, when physicians prescribe the selegiline patch (Emsam) for depression, as long as the dose remains below 12 mg/day, which is standard, it inhibits MAO-B; however, selegiline patch doses greater than 12 mg/day inhibit MAO-A as well as MAO-B, and leave patients at risk of tyramine-induced hypertension.
Dementia
However, as the illness progresses and patients age, dementia routinely complicates the illness. Dementia affects about 20% of all Parkinson disease patients, 40% of those older than 70 years, and more than 80% who have had the illness for 20 years. Its prevalence increases in proportion to the patient’s age, duration of the illness, and physical impairments. Dementia is more frequent when akinesia and rigidity rather than tremor predominate in Parkinson disease. Unlike the dementia in Alzheimer disease, the dementia in Parkinson disease is not associated with apolipoprotein E4 alleles (see Chapter 7).
Parkinson disease dementia, which differs clinically from that of Alzheimer disease dementia, is distinguished by inattention, poor memory, difficulty shifting mental sets, and bradyphrenia (slowed thinking, the cognitive counterpart of bradykinesia), as well as hallucinations. With its almost invariable gait impairment and preserved language function, Parkinson disease dementia serves as a prime example of “subcortical dementia” (see Chapter 7). The Mini-Mental State Examination (MMSE) and the Montreal Cognitive Assessment (MoCA) are each valid screening tests for cognitive impairment in Parkinson disease; however, the MoCA is more sensitive.
A special diagnostic hazard when evaluating a patient with parkinsonism and dementia is failing to recognize dementia with Lewy bodies disease. This illness and Parkinson disease share rigidity and bradykinesia, delusions and hallucinations, and sleep disturbances as well as cognitive impairment. One major clinical difference is that in dementia with Lewy bodies disease, dementia constitutes an initial, not a late-developing, manifestation (see Chapter 7), but it is at least a relatively late development in Parkinson’s disease. On a histologic level, dementia with Lewy bodies disease features Lewy bodies in the cerebral cortex, not just confined to the basal ganglia.
Treatment of Comorbid Psychosis
Because administration of antiparkinson medication before bedtime tends to cause nightmares and hallucinations, neurologists advance the last daily dose to the early evening. Also, they avoid suddenly stopping these medicines because their abrupt withdrawal may lead to irreversible motor deterioration, complications of immobility, or even the neuroleptic-malignant syndrome (NMS) (see Chapter 6).
Pathology of Parkinson Disease
The loss of tyrosine hydroxylase represents the critical failure in Parkinson disease because this enzyme is the rate-limiting enzyme in dopamine synthesis (see Fig. 18-3). With the tyrosine hydroxylase deficit, the ever-shrinking pool of remaining nigrostriatal tract neurons cannot sustain the essential synthetic pathway. Once approximately 30% of these neurons degenerate, the nigrostriatal tract cannot synthesize adequate dopamine and Parkinson disease symptoms appear.
On a microscopic level, neurons in these locations characteristically accumulate Lewy bodies, which contain a core of α-synuclein (see Chapter 7). In contrast, Lewy bodies located in the cerebral cortex, as well as the basal ganglia, constitute the histologic hallmark of dementia with Lewy bodies disease. With their abundance of Lewy bodies, both Parkinson disease and dementia with Lewy bodies disease fall under the rubric of synucleinopathies.
Parkinsonism
Medication-Induced Parkinsonism
When medicines, as opposed to illicit drugs, induce parkinsonism, rigidity is the most prominent feature, all three cardinal features occur in only about one-third of cases, and individuals older than 60 years and women are particularly susceptible. Typical and most atypical antipsychotic agents – because to a greater or lesser degree they block D2 receptors – routinely cause parkinsonism. Tetrabenazine (Xenazine) induces parkinsonism because it depletes dopamine (see later and Figs 18-3 and 18-12). Similarly, nonpsychiatric medicines that block D2 receptors, such as metoclopramide (Reglan), trimethobenzamide (Tigan), prochlorperazine (Compazine), and promethazine (Phenergan), produce the same problem. Case reports have also implicated valproate (Depakote), lithium, amiodarone, and calcium channel blockers – medicines with no direct connection to D2 receptors.
Medication-induced parkinsonism, once physicians discontinue the offending drug, should fully resolve in a few weeks. Nevertheless, signs often persist for 3 months and occasionally for 1 year. However, physicians must be careful with persistent parkinsonism patients because approximately 10% harbor Parkinson disease and some probably have dementia with Lewy bodies disease (see Chapter 7). In children and young adults, persistent parkinsonism following antipsychotic treatment suggests several neurologic illnesses (see later).
Therapy of Parkinson Disease
Medications
L-Dopa.
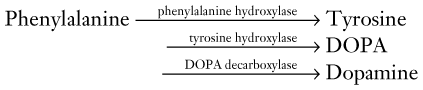
Treatments for Parkinson disease alleviate motor symptoms for many years, but do not reverse the neurodegeneration. Medicines maintain normal dopamine activity by enhancing dopamine synthesis, retarding its metabolism, or acting as agonists at dopamine receptors. Enhancing dopamine synthesis, usually the initial treatment, consists of substituting orally administered L-dopa for endogenous but deficient DOPA in the synthetic pathway (Fig. 18-3).
Dopa- and Dopamine-Preserving Medications
Several medications increase nigrostriatal dopa by inhibiting two enzymes – dopa decarboxylase and catechol-O-methyltransferase (COMT) – that metabolize it in the systemic circulation (Fig. 18-11). With a relative increase in nigrostriatal dopa, nigrostriatal dopamine concentration increases.
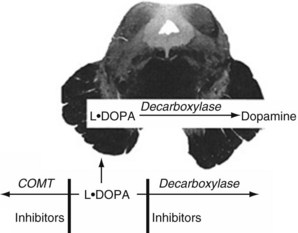
FIGURE 18-11 Medicines that inhibit catechol-O-methyltransferase (COMT), such as entacapone, and those that inhibit decarboxylase, such as carbidopa, slow the metabolism of L-dopa. Because of the blood–brain barrier, they do not affect dopamine synthesis in the nigrostriatal tract. These enzymes, combined with L-dopa, permit smaller L-dopa doses, which minimizes systemic side effects.
On the other hand, although they ameliorate some symptoms and provide a modicum of antidepressant effect, antiparkinson MAO-B inhibitors carry a caveat. At high doses, they inhibit MAO-A as well as MAO-B. Because MAO-A is important in both serotonin and catecholamine metabolism, MAO-B inhibitors place patients in a position where they are vulnerable to the serotonin syndrome or a hypertensive crisis (see previously and Chapters 6, 9, and 21).
Dopamine Agonists
As an initial medication or when dopamine production eventually falls to inadequate levels, dopamine agonists – pramipexole and ropinirole – provide stimulation of postsynaptic dopamine receptors (see Fig. 18-3). Bypassing degenerated presynaptic neurons, dopamine agonists act directly on D2 and, to a lesser extent, on other postsynaptic dopamine receptors.
Other Medications
Anticholinergics reduce tremor in Parkinson disease and other forms of parkinsonism. By reducing cholinergic activity, these medicines seem to act by maintaining the balance with the diminished dopamine activity (Fig. 18-12). On the other hand, anticholinergics routinely produce mental and physical side effects, especially in the elderly.
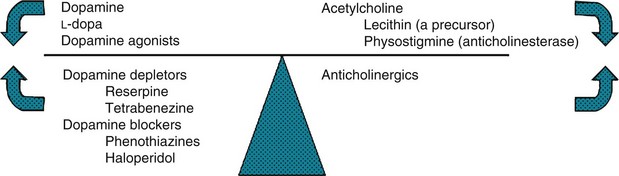
FIGURE 18-12 In a classic but limited model, dopamine and acetylcholine (cholinergic) activity normally balance each other. When Parkinson disease reduces dopamine activity, the left side of the scale rises. Dopamine precursors, dopamine agonists, and anticholinergics – Parkinson disease treatments – restore the balance. Conditions characterized by excessive dopamine activity, such as chorea, push the left side downward. Substances that either antagonize or deplete dopamine, or enhance cholinergic activity, restore the balance.
Athetosis
Athetosis consists of involuntary slow, continually changing, twisting movements predominantly affecting the face, neck, and distal limbs (Fig. 18-13). It sits at the beginning of a sequence – athetosis, choreoathetosis, chorea, and hemiballismus – of progressively larger and more irregular involuntary movements. In a potentially confusing aspect, additional involuntary movements may coexist with athetosis. For example, rapid jerks of chorea may punctuate slow movements of athetosis, and powerful twists of dystonia may interrupt and override athetosis’ slow fluctuations.
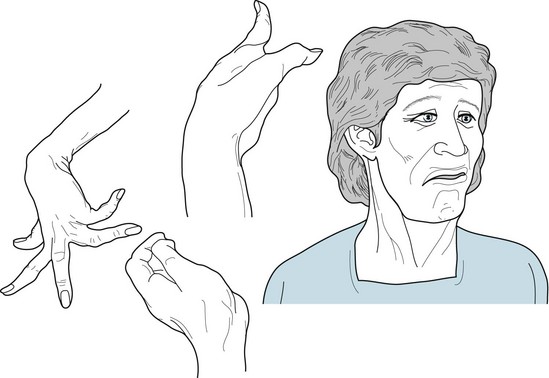
FIGURE 18-13 In athetosis, the face grimaces incessantly. Fragments of smiles alternate with frowns. Pulling of one or the other side distorts the entire appearance. In addition, neck muscles contract and rotate the head. Laryngeal contractions and irregular chest and diaphragm muscle movements cause an irregular speech cadence, nasal pitch, and dysarthria. Fingers writhe constantly and tend to assume hyperextension postures. At the same time, wrists rotate, flex, and extend. Involuntary limb activity prevents writing, buttoning, and other fine tasks, but it usually allows deliberate, larger-scale shoulder, trunk, and hip movement.
Usually encountered as a variety of cerebral palsy (see Chapter 13), athetosis is usually not apparent until early childhood. Most often athetosis results from combinations of perinatal hyperbilirubinemia (kernicterus), hypoxia, and prematurity. Genetic factors are unimportant.
Because athetosis originates in brain injuries that occur during the first 30 days after birth as well as in utero, which neurologists consider congenital brain injuries, seizures and mental retardation frequently accompany this movement disorder. However, with damage confined to the basal ganglia, as in many cases of athetosis, patients have normal intelligence despite disabling movements and garbled speech. Physicians, schoolteachers, family members, and friends should recognize that these patients retain cognitive and emotional capacities despite devastating physical neurologic disabilities.*
Chorea
Huntington Disease
Of the many causes of chorea (Box 18-1), Huntington disease, previously called “Huntington’s chorea,” remains pre-eminent. Chorea, dementia, and behavioral abnormalities characterize this autosomal dominantly inherited disorder. Symptoms first emerge when patients are, on the average, approximately 37 years. However, approximately 10% of patients develop symptoms in childhood or adolescence and 25% when they are older than 50 years. Adults with the illness usually succumb to aspiration and inanition one to two decades after the diagnosis.









