Fig. 2.1
Molecular cascades of calcium and sodium signaling in astroglia. Abbreviations: AMPAR α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor, CBP Ca2+ binding protein, EAAT excitatory amino acid transporter, ER endoplasmic reticulum, GABA γ-aminobutyric acid, GAT GABA transporter, GPCR G-protein coupled receptor, InsP 3 R inositol 1,4,5 trisphosphate (InsP3)-gated Ca2+ channel/receptor, NCX Na+/Ca2+ exchanger, NKA Na+/K+ ATP-ase, NMDAR N-methyl D-aspartate receptor, PMCA plasmalemmal Ca2+-ATPase, P2XR purinergic 2X receptor, SERCA sarco(endoplasmic) reticulum Ca2+ ATPase, SOCE store-operate Ca2+ entry, TRP transient receptor potential. (Modified from Parpura and Verkhratsky 2012)
Activation of metabotropic receptors with subsequent InsP3-induced Ca2+ release from the ER represents the main mechanism of propagating intra- and intercellular Ca2+ waves, the latter commonly considered as a main mechanism for long-range communication in glial syncytia. Propagation of these calcium waves can involve direct diffusion of InsP3 through gap junctions , or astroglial release of neurotransmitters (usually ATP), or combination of both (Giaume and Venance 1998; Scemes and Giaume 2006) . At the same time, the role for another ER Ca2+ channel, the ryanodine receptor (RyR), in glial calcium signaling remains debatable. Astrocytes express RyRs both at the mRNA and protein level (Matyash et al. 2002; Parpura et al. 2011) . Caffeine-induced RyR-mediated Ca2+ release was observed in astrocytes in the thalamus (Parri and Crunelli 2003) , and inhibition of RyRs was shown to reduce ER stress in astrocytes in culture and in organotypic slices (Alberdi et al. 2013) , and yet the physiological role for RyRs in astroglial Ca2+ signaling remains questionable (Beck et al. 2004) .
What are the consequences and the physiological role of ER Ca2+ signaling in astroglia? Several lines of evidence showed that ER Ca2+ release is instrumental for initiating exocytotic neurotransmitter release from astrocytes in vitro (Reyes and Parpura 2009; Parpura et al. 2011) . First, it was demonstrated that the inhibition of Sarco(Endo)plasmic Reticulum Ca2+ ATPases (SERCA) by thapsigargin (that causes depletion of the ER from releasable Ca2+ due to an unopposed leakage) (Fig. 2.1) almost completely blocked the Ca2+-dependent release of glutamate from astrocytes (Innocenti et al. 2000; Jeremic et al. 2001) . Similarly, thapsigargin blocked the mechanically-induced glutamate release from cultured astroglia (Hua et al. 2004) . The same effect was observed after treating astrocytes with diphenylboric acid 2-aminoethyl ester that is known to inhibit InsP3 receptors and store-operated Ca2+ entry (Hua et al. 2004) . Calcium signals originated from the ER initiate the release of vasoactive substances (for example derivatives of arachidonic acid or carbon monoxide) that affect the tone of cerebral arterioles and hence contribute to astroglia-dependent regulation of local blood flow (Zonta et al. 2003b; Mulligan and MacVicar 2004) . There are data that Ca2+ release from the ER regulates astroglial apoptosis through transactivation of pro-apoptotic factor Bax (Morales et al. 2011) . Dynamic changes in ER Ca2+ that accompany Ca2+ release are also involved in the regulation of multiple ER functions, most notably in controlling the activity of chaperones and protein folding; long-lasting decrease in ER Ca2+ level can bring upon ER stress, which contribute to various pathologies (Alberdi et al. 2013) . Finally, global astroglial Ca2+ signals associated with ER Ca2+ release dynamically affect mitochondrial metabolism thus regulating ATP synthesis and providing for activity-metabolic coupling.
At the same time, the role of ER Ca2+ release in astroglial physiology in situ remains debatable. For example, experiments on genetically modified mice in which ER Ca2+ release in astrocytes was specifically affected showed that neither amplification nor occlusion of astroglial metabotropic Ca2+ signaling affects synaptic transmission/plasticity in hippocampus (Fiacco et al. 2007; Petravicz et al. 2008; Agulhon et al. 2010) . Similarly, ultrastructural study has shown that perisynaptic astroglial processes in hippocampus do not contain ER structures, which are localized mainly in the more proximal processes (Partushev et al. 2013) .
These observations revived interest to plasmalemmal Ca2+ fluxes that, in particular, can underlie rapid and highly localized Ca2+ signals in perisynaptic astroglial processes; these Ca2+ signals are critical for the homeostatic control of synaptic cleft and for the regulation of synaptic plasticity.
2.2.2 Plasmalemmal Ca2+ Entry in Astrocytes
Astrocytes have several molecular pathways responsible for the generation of transmembrane Ca2+ fluxes, which include ionotropic receptors , transient receptor potential (TRP) channels, sodium-calcium exchangers (NCX), and possibly voltage-gated Ca2+ channels (Fig. 2.1). The plasma membrane Ca2+-ATPase (PMCA) is the major Ca2+ extruder in resting astrocytes, but it plays a less important role during times of Ca2+cytosolic loads caused by mechanical stimulation (Reyes et al. 2012) .
Ionotropic Receptors
Astrocytes, both in vitro and in situ, are endowed with several types of Ca2+ permeable ligand-gated channels. The first class of these channels is represented by ionotropic glutamate receptors of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) and N-methyl-D-glutamine (NMDA) types; functional expression of kainate receptors in astrocytes has not yet been reported (Parpura and Verkhratsky 2013) . The Ca2+ permeable AMPA receptors (that lack GluA2 subunit) have been routinely found in cultured astrocytes and their expression is confirmed in Bergmann glial cells in the cerebellum (Steinhauser and Gallo 1996) . However, the extent to which Ca2+ permeable receptors are present in other brain regions needs further characterization. Nonetheless, Ca2+ permeability of GluA2-devoid AMPA receptors is relatively low (PCa/Pmonovalent ~ 1, (Burnashev et al. 1992)) which, together with rapid desensitization of AMPA receptors in physiological context, very much limits Ca2+ entry. In contrast, NMDA receptors, that have been found in mouse cortical astrocytes and also identified in human astroglia (Lalo et al. 2006; Verkhratsky and Kirchhoff 2007; Lee et al. 2010; Lalo et al. 2011) , are characterized by much larger Ca2+ permeability (PCa/Pmonovalent ~ 3) and slow desensitization that allows Ca2+ influx, resulting in substantial [Ca2+]i rise in astrocytes studied in acute isolation and in acute brain slices (Palygin et al. 2010) . In addition, astroglial NMDA receptors have a weak Mg2+ block at physiological resting potential that permits receptor activation by glutamate release during on-going synaptic activity (Lalo et al. 2011) . Astroglial Ca2+ signaling is also mediated by ionotropic P2X purinoceptors; specific heteromeric P2X receptors are operative in neocortical astroglia (Lalo et al. 2008). These receptors have very high sensitivity to ATP (EC50 ~ 50 nM) and do not desensitize in the presence of an agonist. These receptors are Ca2+ permeable (PCa/Pmonovalent ~ 2) and, similar to NMDA receptors, mediate [Ca2+]i signals in astrocytes in isolation and in situ (Palygin et al. 2010) . Astrocytes in the neocortex were also reported to express P2X7 receptors which, when activated, may provide large Ca2+ influx (Norenberg et al. 2010; Oliveira et al. 2011) , although these receptors, most likely, mediate pathological responses associated with massive release of ATP (Franke et al. 2012; Illes et al. 2012) . Besides ionotropic glutamate receptors and purinoceptors mediated Ca2+ entry into astrocytes, these ions can enter through α7 Ca2+ permeable nicotinic cholinoreceptors, identified in cultured astroglia (Sharma and Vijayaraghavan 2001; Oikawa et al. 2005) .
TRP Channels
Similarly, Orai channels and their respective currents have been recently recorded in primary cultured astrocytes and astroglial cell lines, which also expressed stromal interacting molecule1 (STIM1), the molecular sensor that monitors the intra-ER Ca2+ concentration (Moreno et al. 2012; Motiani et al. 2013) .
Astrocytes express TRPC1,4,5 subunits at both mRNA and protein levels (Pizzo et al. 2001; Grimaldi et al. 2003; Golovina 2005; Malarkey et al. 2008) . In TRPC heteromultimers the TRPC1 channel is obligatory subunit, whereas TRPC4 and TRPC5 proteins have an auxiliary role (Strubing et al. 2001; Hofmann et al. 2002) . Specific inhibition of TRPC1 channels by either expression down-regulation with an antisense or by exposing cells to a blocking antibody directed at an epitope in the pore forming region of the TRPC1 protein substantially reduced SOCE following metabotropic or mechanical stimulation in cultured astrocytes (Golovina 2005; Malarkey et al. 2008) .
Hippocampal astrocytes seem to express TRPA1 mediating “spotty”, localized near-membrane, spontaneous [Ca2+]i changes (Shigetomi et al. 2012) . Hence, in cultured astrocytes, these [Ca2+]i transient were inhibited by the broad spectrum TRP channel antagonist HC 030031 and by anti-TRP silencing RNA, while the TRPA1 agonist allyl isothiocyanate increased frequency of these events. Activity of TRPA1 channels contributed to setting the resting [Ca2+]i in hippocampal astrocytes (both in culture and in situ), as inhibition of these channels resulted in a significant (from ~ 120 nM to ~ 50 nM) decrease in basal [Ca2+]i (Shigetomi et al. 2012) .
Sodium/Calcium Exchanger
Important molecular pathway regulating Ca2+ entry especially in astroglial perisynaptic processes is represented by sodium-calcium exchange mechanism . Astrocytes are in possession of all three types of mammalian Na+/Ca2+ exchangers, the NCX1, NCX2 and NCX3. The NCX molecules are concentrated in the perisynaptic processes and are often co-localized with plasmalemmal glutamate transporters and NMDA receptors (Minelli et al. 2007) . The NCX dependent Ca2+ transport in astrocytes operates in both forward (Ca2+ extrusion) and reverse (Ca2+ entry) modes; because of the relatively high cytosolic concentration of Na+ ions (see below), the reversal potential of NCX is set rather close to astroglial resting membrane potential and therefore even a moderate depolarization or an increase in the intracellular Na+ readily reverses the NCX and favors Ca2+ influx (Kirischuk et al. 2012) . The NCX-mediated Ca2+ fluxes in both forward and reverse modes are documented for primary cultured astrocytes and astroglial cells in situ (Goldman et al. 1994; Kirischuk et al. 1997) . Influx of Ca2+ through NCX can be activated by cytosolic Na+ increase following the activation of ionotropic receptors (Kirischuk et al. 1997) or glutamate transporters (Kirischuk et al. 2007) ; in cultured astrocytes the reverse mode of NCX can be induced by a moderate depolarization (Paluzzi et al. 2007) .
Voltage-gated Ca2+ Channels (VGCCs)
Although there are numerous reports indicating the expression of VGCCs in astrocytes in vitro (see (Parpura et al. 2011; Verkhratsky et al. 2012) for details and references), the role for these channels in physiologically relevant Ca2+ signaling in astroglia in the brain tissue remains questionable. There are some indications for VGCC-dependent Ca2+ dynamics in astrocytes from the neurogenic subventricular zone (Young et al. 2010) and the ventrobasal thalamus (Parri et al. 2001; Parri and Crunelli 2003) and yet these reports remain sporadic and unconfirmed. It might be argued that VGCCs may become important for Ca2+ signals in reactively remodeled astroglia. In particular, reactive astrocytes in the hippocampi of young mice, which experienced pilocarpine-induced status epilepticus, showed an increased expression of L- and P/Q- type channels at 1 week and 2 months following an insult (Xu et al. 2007) .
2.2.3 Mitochondria in Astroglial Ca2+ Signaling
Mitochondria are able to accumulate Ca2+ ions from the cytosol through Ca2+ ion channels localized in the outer and in the inner membrane; these channels are represented, respectively, by the voltage-dependent anion channels with considerable Ca2+ permeability and by the highly selective Ca2+ uniporter (composed of the channel protein of mitochondrial calcium uniporter, and auxiliary EF-hand-containing protein that regulates the uniporter, MICU1/CBARA1 (De Stefani et al. 2011)) . Mitochondrial Ca2+ sequestration has been well documented in astrocytes (Reyes and Parpura 2008) . In addition, mitochondria may release Ca2+ via mitochondrial Na+/Ca2+ exchanger NCLX (Parnis et al. 2013) and through the mitochondrial permeability transition pore (Basso et al. 2005; Reyes and Parpura 2008) .
2.3 Glial Sodium Signaling
2.3.1 Molecular Physiology of Na+ Signaling
Many astroglial functions are regulated by the transmembrane gradient for Na+ ions (Verkhratsky et al. 2013c) , which in turn is subject to dynamic variations induced by the physiological stimulation of astrocytes. Astroglial sodium homeostasis is somewhat different from neurons at least in one parameter: the resting cytosolic Na+ concentration ([Na+]i) in astrocytes is about two times higher (Rose and Ransom 1996; Reyes et al. 2012; Unichenko et al. 2012) . High levels of [Na+]i are important because they set reversal potentials for numerous Na+-dependent solute transporters expressed in astroglial membrane (Parpura and Verkhratsky 2012) . Physiological stimulation of astrocytes in vitro or in situ by exogenous neurotransmitters, by synaptic inputs, or by mechanical indentation trigger transient elevation of [Na+]i by up to 20–25 mM from the resting level. Furthermore, these [Na+]i increases may propagate through astroglial syncytia using gap junctions , thus, generating propagating Na+ waves. These observations led to a concept of astroglial Na+ signaling (see (Kirischuk et al. 2012; Langer et al. 2012; Parpura and Verkhratsky 2012; Rose and Karus 2013; Verkhratsky et al. 2013c) and references therein) .
Increases in [Na+]i are mediated through several molecular pathways (Fig. 2.1). Sodium fluxes are generated by all ionotropic receptors present in astroglial membranes, these receptors being, in essence, cationic channels provide for a large Na+ entry upon their activation (Kirischuk et al. 1997; Langer and Rose 2009) . Another important route is associated with the activation of TRPC channels (Reyes et al. 2013) , which (through store-operated mechanism) provide a link between ER Ca2+ release and plasmalemmal Na+ flux (Verkhratsky et al. 2013b). In astrocytes from subfornical organ Na+ influx is mediated by Nax channels sensitive to extracellular Na+ concentration. Opening of these channels with subsequent Na+ entry is instrumental for astroglial chemosensing and systemic regulation of Na+ homeostasis (Shimizu et al. 2007) .
Another physiologically important Na+ influx pathway is associated with the activation of neurotransmitter transporters for glutamate and GABA , the astroglial specific excitatory amino acid transporters 1 and 2 (EAAT1 and EAAT2) and the GABA transporters of GAT1 and GAT3 types. The stoichiometry of EAAT1/2 is 1 Glu−:3 Na+:1K+:1H+, of which Na+, proton and glutamate enter the cell in exchange to K+ efflux, whereas GAT1 and GAT3 exchange 1 GABA molecule for 2 Na+ ions and 1 Cl− anion. Accordingly, in physiological conditions, astroglial uptake of neurotransmitters is accompanied with net Na+ influx that underlie electrogenicity of transporters and can increase [Na+]i by ~ 20 mM following the activation of EAAT1/2 or by ~ 7 mM following the activation of GAT1/3 (Kirischuk et al. 2007; Unichenko et al. 2012) .
Astrocytes possess Na+/K+-ATPase (NKA), a pump which is the major Na+ extruder in resting astrocytes. However, NKA seems to be less important during times of Na+ cytosolic loads caused by mechanical stimulation. Instead, NCX operating in reverse mode appeared as a major contributor to the overall Na+ homeostasis in astrocytes, both at rest and when these glial cells were mechanically stimulated (Reyes et al. 2012) .
2.3.2 Functional Role of Na+ Signaling
Increases in [Na+]i regulate multiple molecular cascades responsible for astroglial homeostatic function (Fig. 2.2). Elevation in [Na+]i activates astroglial NKA, which (i) affects K+ buffering (Wang et al. 2012) and (ii) triggers lactate synthesis and therefore mobilize astrocyte-neuron lactate shuttle (Pellerin and Magistretti 2012) ; lactate can be released (or taken up) from astrocytes through the proton-coupled transporter MCT-1. In this way astroglial metabolic support is tailored to the neuronal activity and hence local neuronal energy demands. Changes in [Na+]i also directly affect Kir4.1 channels that are critical for astroglial K+ buffering (Kucheryavykh et al. 2012) , and regulate other K+ transporters such as, for example, Na+/K+/Cl− co-transporter NKCC1, which also contributes to K+ buffering (Kirischuk et al. 2012) .
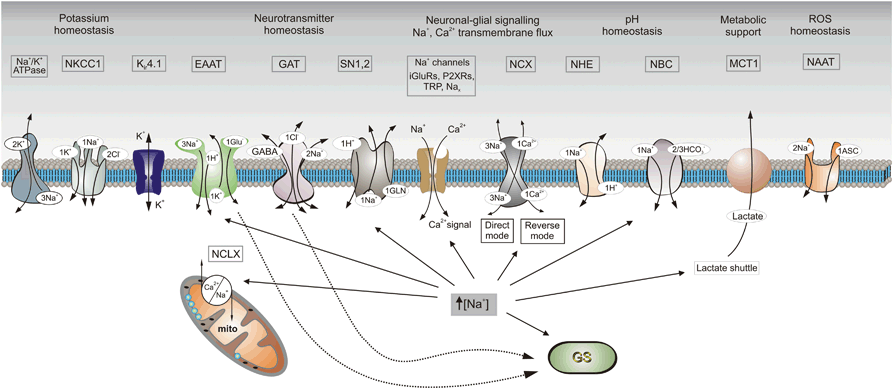
Fig. 2.2
Functional targets of Na+ signaling in astroglia. Schematic diagram showing receptors and transporters involved in and sensitive to changes in [Na+]i and their relations to main homeostatic functions of astroglia. Abbreviations: EAAT excitatory amino acid transporters, GAT GABA transporters, GS glutamine synthetase, iGluRs ionotropic glutamate receptors, mito mitochondrion, Kir4.1 inwardly rectifying K+ channels involved in K+ buffering, MCT1 monocarboxylase transporter 1, Na x Na+ channels activated by extracellular Na+, NAAT Na+-dependent ascorbic acid transporter, NBC Na+/HCO3 − (sodium-bicarbonate) co-transporter, NCX Na+/Ca2+ exchanger, NCLX mitochondrial Na+/Ca2+ exchanger, NHE Na+/H+ exchanger, NKCC1 Na+/K+/Cl− cotransporter, P2XRs ionotropic purinoceptors, SN1,2 sodium-coupled neutral amino acid transporters 1 and 2 which underlie the extrusion of glutamine, TRP transient receptor potential channels. (Modified and updated from Kirischuk et al. 2012)
Changes in [Na+]i have profound effects on many astroglial solute carriers that are controlling molecular homeostasis of the CNS environment. First and foremost, [Na+]i regulates neurotransmitter homeostasis. Both glutamate and GABA plasmalemmal transporters are directly affected by [Na+]i, albeit with different functional consequences. Physiological increases in [Na+]i may slow down, but never reverse glutamate transporters , which have reversal potential ~ + 60 mV (Kirischuk et al. 2007, 2012) . Reversal of glutamate transporter, that has been observed in experiment (Szatkowski et al. 1990) , may only occur in pathology when massive [Na+]i increase coincides with an increase in cytosolic glutamate concentration and membrane depolarization . In contrast, reversal potential of astroglial GABA transport (~ − 80 mV) is quite close to the resting membrane potential, and therefore even minute increases in [Na+]i (~ 7 mM) can switch GABA transport into the reverse mode and, thus, mediate GABA release from astroglia (Unichenko et al. 2012) . Cytosolic Na+ levels also regulate glutamine synthesis and glutamine release by directly affecting glutamine synthetase and the plasmalemmal sodium-dependent neutral amino acid transporter SNAT3/SLC38A3, respectively (Mackenzie and Erickson 2004) . In this way astroglial Na+ signaling regulates both excitatory and inhibitory neurotransmission, because both are critically dependent on continuous supply of glutamine acting as a precursor for glutamate and GABA.
Astroglial [Na+]i increases favor reversal of NCX, which, in astrocytes , dynamically fluctuates between forward/reverse modes and thus mediates local Ca2+ signals. Similarly [Na+]i levels regulate mitochondrial exchanger NCLX, thus, controlling mitochondrial Ca2+ release (Parnis et al. 2013) . Astroglial Na+ signals also modulate several other homeostatic pathways, such as Na+/H+ exchanger (NHE), which regulate pH homeostasis, and sodium-bicarbonate co-transporter (NBC). Intracellular Na+ also controls the release of radical oxygen species (ROS) scavenger ascorbic acid through dedicated Na+-dependent transporter NAAT (see (Kirischuk et al. 2012; Parpura and Verkhratsky 2012; Verkhratsky et al. 2013c) for details) .
2.4 Glial Ca2+ Signaling in Pathology
Astroglial contribution to neurological diseases is fundamental for the progression and outcome of pathology; this contribution includes neuroprotection through the activation of astroglial homeostatic cascades and through the induction of reactive astrogliosis (Verkhratsky et al. 2013a).
Focal Ischemia and Stroke
Astrocytes are intimately involved in the pathogenesis of ischemic insults to the CNS. Cessation of blood flow triggers necrotic process in the infarction core, where all cells, including astroglia, die; this process results from a failure of ion homeostasis and overloading the cells with Ca2+ and Na+. In the ischemic penumbra, surrounding the core region, the cellular fate can be different, and indeed the progression of cell death through the penumbra that may take days after the initial insult, is to a large extend, defined by astrocytes (Nedergaard and Dirnagl 2005; Giaume et al. 2007) . In the penumbra region, astrocytes display higher resistance compared to all other cell types; they not only outlive neurons but can support them through the release of neurotrophic factors and ROS scavengers, as well as through containing glutamate excitotoxicity , K+ buffering and metabolic support (Tanaka et al. 1999; Pekny and Nilsson 2005; Verkhratsky et al. 2013a) . Most of these neuroprotective functions are regulated by astroglial ionic signaling. At the same time astrocytes may contribute to the spread of death signals through the penumbra; this spread can be associated with aberrant astroglial Ca2+ waves that can produce glutamate release distal to the infarction core and, thus, induce distant cell death (Nedergaard and Dirnagl 2005; Takano et al. 2009) . These aberrant glial Ca2+ signaling can be further exacerbated by the ATP released from the damaged cells; high concentration of ATP activates both metabotropic and P2X7 ionotropic astroglial receptors that results in pathological Ca2+ signaling (Franke et al. 2012) .
Epilepsy
Astroglial contribution to the pathogenesis of epilepsy received increasing attention in recent years . Profound astrogliosis is regularly observed in both animal models of the disease and in human brain tissue (de Lanerolle and Lee 2005; Seifert et al. 2006) ; these changes developing in parallel with functional astroglial deficiency that includes impaired K+ buffering, neurotransmitter uptake and water transport (Seifert et al. 2006). Pathological ionic signaling in astrocytes can even contribute to epileptogenesis. Aberrant Ca2+ signaling manifested by spontaneous Ca2+ oscillations was observed in astrocytes isolated from the brains of patients suffering from Rasmussen’s encephalitis, the autoimmune form of child epilepsy. These abnormal Ca2+ signals are arguably connected with pathological modification of AMPA receptors that results in their reduced desensitization and increased Ca2+ influx (Manning and Sontheimer 1997; Seifert et al. 2004) . Astroglial Ca2+ signaling can also be affected through an increase in the expression of metabotropic glutamate receptors and InsP3-induced Ca2+ release. Pathologically, increased astroglial Ca2+ signals may contribute to the formation of a recurrent excitatory loop with neurons, thus, initiating a focal seizure (Carmignoto and Haydon 2012) . Finally, aberrant astroglial Ca2+ signals may induce a massive release of glutamate that could trigger paroxysmal depolarization shifts in neurons, thus, provoking seizure (Tian et al. 2005) . In addition, the epileptic tissue is characterized by impaired inhibitory transmission and it appeared that the GABA-ergic transmission is particularly sensitive to astroglial glutamine supply; epilepsy is accompanied with an inhibition of glutamine synthetase that suppresses GABA-ergic inhibitory transmission (Ortinski et al. 2010) .
Psychiatric Disorders
Related posts:
 General Pathophysiology of Neuroglia: Neurological and Psychiatric Disorders as Gliopathies
General Pathophysiology of Neuroglia: Neurological and Psychiatric Disorders as Gliopathies
 Emerging Roles for Glial Pathology in Addiction
Emerging Roles for Glial Pathology in Addiction
 Enteric Glial Cells: Implications in Gut Pathology
Enteric Glial Cells: Implications in Gut Pathology
 Astroglial Connexins as Elements of Sleep-Wake Cycle Regulation and Dysfunction
Astroglial Connexins as Elements of Sleep-Wake Cycle Regulation and Dysfunction
 Novel Therapeutic Approaches to Malignant Gliomas
Novel Therapeutic Approaches to Malignant Gliomas
 Neurodegeneration and Neuroglia: Emphasis on Astroglia in Alzheimer’s Disease
Neurodegeneration and Neuroglia: Emphasis on Astroglia in Alzheimer’s Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


