Typical brain MRI in a patient with sJCD. FLAIR (top row), DWI (middle row), and ADC map (bottom row) sequences show the typical T2-weighted hyperintensities with corresponding restricted diffusion. as well as the striatum and/or thalamus. This MRI shows restricted diffusion (bright on FLAIR/DWI and dark on ADC map) of frontal, parietal, and cingulate cortices (“cortical ribboning”), bilateral striatum, and lateral thalamus. Note that the hyperintensities are seen much better on DWI than on FLAIR, which is typical of most cases of sJCD.
Each of the original six molecular subtypes of sJCD described above has been associated with certain MRI DWI patterns [46] (see Table 21.1). In our experience, MRI DWI/ADC signal changes in sJCD are solely neocortical in about 24–30%, both neocortical and subcortical in ~68%, or predominantly subcortical (mostly involving the striatum, with or without thalamic involvement) in about 2–5% of cases [70, 74]. We have found that upwards of 90% of sJCD cases demonstrate DWI signal changes in limbic and paralimbic cortical structures; isolated limbic involvement, however, is atypical for sJCD and should alert clinicians towards alternative diagnoses, such as autoimmune encephalitis or seizures [70]. Cortical ribboning in sJCD can be unilateral or bilateral, but typically spares the precentral cortex [70]. There is often an anterior to posterior decreasing gradient in the DWI or T2-weighted signal intensity of the striatum (i.e., the anterior caudate appears more hyperintense than the posterior putamen) [70]. Some MRI criteria consider T2 or fluid attentuated inversion recovery (FLAIR) MRI abnormalities to be sufficient for sJCD diagnosis [75], but we and others think that these sequences are inadequate due to poor sensitivity and that identifying abnormalities on DWI, ideally with restricted diffusion confirmed on ADC map, is necessary [70, 76].
It is important to stress that cortical ribboning and/or deep nuclei hyperintensity sometimes are seen in other conditions, such as viral and/or autoimmune encephalitis, hypoglycemia, seizures and status epilepticus, hyperammonemic encephalopathy, hypoxic injury, mitochondrial encephalopathy, vasculitis, extrapontine myelinolisis, Wilson’s disease, Wernicke encephalopathy, and hyperglycemia with seizures [70, 71, 77, 78].
Electroencephalography (EEG). The presence of periodic sharp wave complexes (PSWCs) on EEG, often biphasic is triphasic and occurring every 0.5–2 seconds, is considered virtually pathognomonic of sJCD in the right clinical setting. This finding only has about 66% sensitivity, however [33, 79], and the presence of PSWCs can be seen rarely in other neurological conditions such as Alzheimer’s disease, dementia with Lewy bodies, toxic–metabolic and anoxic encephalopathies (e.g., hepatic), progressive multifocal leukoencephalopathy, and Hashimoto’s encephalopathy [80, 81].
Cerebrospinal fluid (CSF). Routine CSF analysis is typically unrevealing in sJCD, except for mildly elevated protein (typically less than 75 mg/dl). Pleocytosis (> 10 WBC cells), elevated IgG index, and/or the presence of oligoclonal bands can occur but are unusual and should alert the clinician to consider alternative diagnoses [82, 83].
Four CSF proteins – tau, 14–3-3, astrocytic protein S100β, and neuron specific enolase (NSE) – commonly are used to aid in the diagnosis of sJCD in clinical and research settings. The sensitivity and specificity of these markers, and therefore their clinical diagnostic utility, is debatable. For example, the nearly perfect sensitivity and specificity ascribed to protein 14–3-3 in early studies [84] decreased considerably in later, larger studies [85–87], the most recent of which reported a sensitivity of 86% and specificity of 74% in pathologically confirmed cohorts [87, 88]. Similarly, the sensitivity and specificity of total-tau (t-tau), NSE, and the astrocytic protein S100β vary greatly among studies [86, 89, 90]. Recently, an elevation of the t-tau/p-tau (phosphorylated tau) ratio was shown to be useful in separating sJCD from other neurodegenerative disorders [91].
The variable sensitivity and specificity of these CSF proteins are probably a reflection of the fact that none of them is intrinsic to prion pathophysiology. Instead, these proteins are part of the normal brain parenchyma and are released into the CSF as a result of various types of tissue injury. Protein 14–3-3, for example, has been shown to be elevated in various disorders such as multiple sclerosis, acute stroke, HIV-related neurodegeneration, and Alzheimer’s disease, among others [92–95]. Hence, the American Academy of Neurology suggests ordering CSF 14–3-3 only when there is a strong clinical suspicion of sJCD, but diagnosis remains uncertain [96].
In our center, we find t-tau as an overall better biomarker for sJCD than 14–3-3 or NSE, although NSE has somewhat higher specificity. Nevertheless, we routinely send all three biomarkers in suspected cases of sJCD as a general screen of rapid neuronal injury and in some cases when MRI is negative or unclear they might be helpful.
A more recent CSF diagnostic test, the “RT-QuIC” (Real-Time Quaking-Induced Conversion assay) is available clinically in some countries; it relies on the detection of actual PrPSc by an amplification method [97]. Although the sensitivity of this test is still low, about 70%, its specificity has been reported to be as high as 98% [98, 99], although some false positive results have been reported. A recent study showed the RT-QuIC assay that reliably detect PrPSc in nasal brushings of psymptomatic patients [98].
Variably protease-sensitive prionopathy (VPSPr)
PrPSc from patients with sJCD largely resists degradation by protease enzymes, such as proteinase K. When protease-treated PrPSc is run on a Western blot, three bands corresponding to mono-, di-, and unglycosylated forms of the protease resistant portion of the protein usually are seen. In 2008, a form of sporadic prion disease was reported in which PrPSc was unusually sensitive to protease and instead of three discrete bands being present on Western blot, there was a ladder of mostly smaller bands. This form of prion disease was named variably protease sensitive prionopathy (VPSPr) [100].
As of 2014, approximately 34 cases of VPSPr have been identified worldwide [41, 101]. Initial psychiatric manifestations are common, often followed by cognitive decline (mostly aphasia) and motor impairment (ataxia and parkinsonism) [41, 102]. Although based on a limited number of cases, PRNP codon 129 polymorphisms appear to affect the clinical phenotype of VPSPr [41]; patients with codon 129MM often have less prominent psychiatric features, whereas those with 129MV and 129VV seem to lack parkinsonism and ataxia. The distribution of codon 129 polymorphism is almost the opposite of typical sJCD, with 62% being VV, 26% MV, and only 12% MM. Survival with VPSPr appears to be longer than patients with classic sJCD, ranging from 18 to 41 months, depending on codon 129 polymorphism [41]. The diagnostic utility of CSF biomarkers, EEG, and DWI MRI for VPSPr is unknown, but in our limited experience with this subtype, MRIs often do not show the classic cortical or deep nuclei T2 or DWI hyperintensities that are seen in sJCD [41], and even when they are present can be subtle (Geschwind et al., unpublished data).
Genetic human prion disease
Historically, prior to the discovery of the PRNP gene, familial forms of PrD were divided into three main phenotypes, based on clinical and pathological characteristics: Gerstmann–Sträussler–Scheinker disease (GSS), familial Jakob–Creutzfeldt disease (fJCD), and fatal familial insomnia (FFI) [104]. Although these syndromic diagnoses are still used clinically today, and will be described briefly in this section, it is important to mention that the spectrum of clinical syndromes seen in familial PrD has widened significantly after the discovery of the PRNP gene and other related genes and polymorphisms [105]. Therefore, clinicians should not discard the possibility of an inherited form of PrD in patients that do not fit the three classic syndromes described below.
All familial or genetic human PrDs (gPrDs) are associated with autosomal dominant mutations of the PRNP gene [104, 106]. Most of the approximately 30 disease-associated PRNP mutations are nearly 100% penetrant; a notable exception is the E200K mutation, which has variable penetrance. Still, clinicians should be aware that a negative family history is not uncommon in patients with gPrD; in one study, up to 47% of patients with gPrD were reported to have no family history of hPrD or other neurological disorders [107].
Familial Jakob–Creutzfeldt disease (fJCD)
Most disease-associated mutations of PRNP lead to fJCD, which may be clinically indistinguishable from sJCD. Patients with fJCD typically present with rapidly progressive dementia, ataxia, and other motor manifestations; onset of disease usually occurs between ages 30 and 55 years [104]. By far, the most common PRNP mutation associated with fJCD and the most common PRNP mutation is E200K [106], which is most common among Libyan Jews and Slovakians.
Patients with fJCD typically present with MRI features that may be indistinguishable from sJCD, which in turn vary according to mutation type. For example, fJCD due to E200K mutations typically shows symmetric, prominent striatal T2-weighted/DWI hyperintensities, often with less prominent cortical ribboning [108], whereas patients with V180I mutations often show cortical ribboning with minimal or absent basal ganglia hyperintensities [109]. EEG may also vary according to mutation subtype, but in general PSWCs tend to appear late. The CSF biomarkers used to evaluate sJCD (protein 14–3-3, NSE, tau) are often elevated in fJCD as well, but these typically offer lower sensitivity than in sJCD [86].
Gerstmann–Sträussler–Scheinker syndrome (GSS)
Patients with GSS tend to become symptomatic in their 50s or younger, usually earlier than patients with sJCD. Motor impairment (ataxia, parkinsonism, etc.) is a common initial manifestation of classic GSS, whereas cognitive impairment tends to occur later [104]. In a subset of patients, psychiatric manifestations may occur early in the disease course [110].
Of note, large pedigree studies have shown that there is considerable phenotypic variability between family members carrying a GSS-associated PRNP mutation, owing to codon 129 polymorphisms [111].
Brain MRI, EEG studies, and CSF investigations are usually unhelpful in “ruling in” the disease. EEG in patients with GSS usually reveals non-specific slow waves, CSF biomarkers (protein 14–3-3, NSE, and tau) are typically not elevated [86], and brain MRI does not demonstrate a reliable pattern of specific abnormalities in this disorder [70].
Fatal familial insomnia (FFI)
FFI is a very rare form of gPrD associated with a specific PRNP point mutation, D178N. Disease onset is typically in the late 40s, and patients usually present with severe progressive insomnia over several months, followed by severe dysautonomia (e.g., tachycardia, hyperhydrosis, and hyperpyrexia). Motor and cognitive manifestations tend to occur later. Mean survival is about 18 months , which is slightly longer than most sJCD patients [33, 104]. In patients with the D178N mutation, the cis codon 129 strongly affects the phenotype. Patients with cis methenione at codon 129 typically present as FFI, whereas those with valine at codon 129 usually present as JCD although in reality patients present along a spectrom [112]. The effect of the codon 129 polymorphism on the D178N phenotype is not absolute, as JCD and FFI can both occur in the same family [113].
Similarly to patients with GSS, EEGs in patients with FFI usually show generalized slowing, without PSWCs. Brain MRI and CSF biomarkers are equally unhelpful [86]. FDG-PET imaging, on the other hand, may reveal thalamic and cingulate hypometabolism [114].
Acquired forms of human prion disease
Acquired forms of hPrD are very rare and fortunately declining in incidence, therefore they will not be discussed in detail in this chapter. Kuru has been effectively eradicated due to public health campaigns that eliminated the practice of endocannibalism among the Fore people of Papua New Guinea. Hence, this disorder will not be discussed.
Variant JCD (vJCD)
First identified in the United Kingdom (UK) in 1995 [115], vJCD represents the only form of hPrD known to be transmitted directly from animals to humans. The vJCD endemic began after more than 180,000 cattle around the world, and especially those within the UK, were fed scrapie-infected sheep products [115]. A subset of these cattle developed bovine spongiform encephalopathy (BSE), or “mad cow disease,” an animal form of PrD. Food products derived from these infected cattle were consumed by humans, a portion of whom developed vJCD. The BSE endemic peaked around 1993, and was controlled by the late 1990s, early 2000s [116]. The vJCD form peaked in 2000 (Figure 21.2), and very few cases have been reported after 2012. As of early 2016, there have been a total of about 230 cases of vJCD reported worldwide. Most cases of vJCD (174) have occurred in the UK and France [27]. Several cases reported in other countries likely had their exposure while residing in the UK [117]. Four cases have been identified in the USA and two in Canada, all of whom acquired the disease in the UK or Gulf States [117, 118].
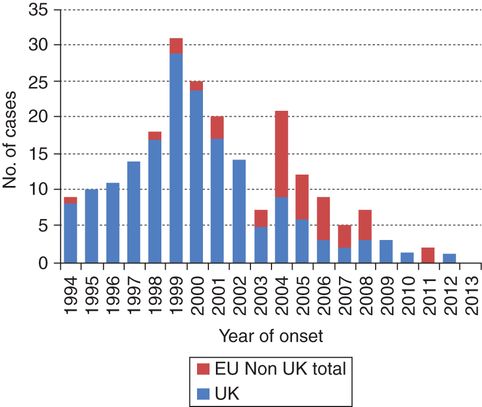
Number of reported vJCD cases (probable and definite) by year of onset in the EU by reporting country, 1994–2014. In 1999 the epidemic of vJCD peaked in the UK, declining progressively with only one probable incident case in 2012, diagnosed and notified in 2013 (as of 1 January 2015). There is a clear delay between the UK epidemic and the occurrence of cases in other EU countries, which peaked in 2004, with 11 incident cases. The most affected country, besides UK, was France with a total of 27 cases. See more at: http://ecdc.europa.eu/en/healthtopics/Variant_Creutzfeldt–Jakob_disease%28vCJD%29/Pages/factsheet_health_professionals.aspx?preview=yes&pdf=yes#sthash.l95bLRSX.dpuf.
Despite successful reduction of the vJCD endemic, there remains the risk of acquiring vJCD via the transfusion of blood products derived from vJCD-infected donors. At least three such cases have been reported, with symptom onset dating 6.5 to 7.8 years after transfusion. The donors were asymptomatic and several years from onset of vJCD when their blood was infective [119, 120]. Furthermore, based on anonymous tonsil and appendix surveys in the UK, upwards of 1 in 2,000 persons in the UK might be harboring latent vJCD and thus might eventually develop the disease or be capable of transmitting it [121–123].
The clinical manifestations of vJCD are typically less protean than in sJCD. Most patients present with psychiatric symptomatology, which may appear up to 6 months before the onset of traditional neurological impairment [124]. Dysesthesias, cerebellar dysfunction, and involuntary movements such as dystonia, myoclonus, and chorea usually appear several months after psychiatric onset [124, 125]. The median age of onset of patients with vJCD is roughly 27 years (range 12–74 years), median disease duration is 14.5 months [124] (longer than patients with sJCD). Nearly every reported case of symptomatic vJCD is homozygous for methionine at codon 129 of the PRNP gene, suggesting this is a susceptibility factor for the disease [126].
The “pulvinar sign” on brain MRI, in which the pulvinar of the posterior thalamus is brighter than the anterior putamen (on T2-weighted sequences), is considered highly supportive for vJCD in the right clinical setting. The pulvinar sign is seen in up to 85% of patients with vJCD. This finding is rarely seen in other forms of hPrD, particularly sJCD [127, 128]. CSF and EEG studies are less sensitive than in sJCD [129, 130]. Tonsillar biopsy also can be useful in the diagnosis of vJCD [131].
Iatrogenic JCD (iJCD)
Aside from kuru, human-to-human transmission of prion disease has only occurred iatrogenically through infected corneal transplants, EEG depth electrodes, cadaveric human pituitary hormone extracts, packed red blood cells, and dura mater graft transplants [10, 132, 133]. Most of these forms of transmission are exceedingly rare, and will not be discussed in detail in this chapter. New cases of iJCD acquired from prior exposure to infected human growth hormone (hGH) extracts and dura mater (DM) grafts, as well as from vJCD blood as noted above, are likely to be identified, however.
Approximately 226 cases of hGH-associated iJCD cases have been reported throughout the world, mostly in patients who received cadaveric hGH from the USA National Hormone and Pituitary Program (NHPP) prior to 1977 (approximately 2,700 recipients) [134], but at least two cases from commercial (private) programs [135]. Cases continue to be identified from exposure two to three decades ago. In Europe, however, cases continue to appear at a rate of approximately two per year, with the most recent case to our knowledge reported in 2011 [134]. The incubation period for hGH-associated iJCD varies widely, from 5 to 42 years (mean 17 years) [134].
Clinically, patients with hGH-associated iJCD usually present with prominent cerebellar signs, with cognitive dysfunction occurring later. Brain MRI findings in hGH-associated iJCD have not been thoroughly studied; some cases show isolated DWI/ADC cerebellar involvement [136], whereas others present similarly to sJCD [135]. Methionine homozygosity has been found to be present in 55% of affected US and French individuals (compared to 40% methionine homozygosity in the general population), suggesting this is a genetic susceptibility factor.
DM-associated iJCD is as prevalent as hGH-associated iJCD. Approximately 228 cases of DM-associated iJCD have been reported, 142 in Japan, although none have been reported in the USA [132, 137]. Most cases have been linked to grafts manufactured by a German company, which marketed dura mater grafts globally in the 1980s. Patients received these grafts until 1993 [132]. Because the mean incubation period of DM-associated iJCD is 12 years (16 months to more than 30 years) [132], new cases are possible. The clinical phenotype of DM-associated iJCD is mostly similar to that of sJCD [132].
The incidence of hGH- and DM-associated iJCD has declined steadily since peaking in the 1990s thanks to the introduction of recombinant growth hormone extracts and synthetic dura mater grafts [33]. Still, given the long incubation times reported for both modes of transmission, additional cases may be diagnosed in the future.
References:
Related posts:
 The non-fluent/agrammatic variant of primary progressive aphasia
The cognitive neurology of corticobasal degeneration and progressive supranuclear palsy
The non-fluent/agrammatic variant of primary progressive aphasia
The cognitive neurology of corticobasal degeneration and progressive supranuclear palsy
 Amnestic mild cognitive impairment
Amnestic mild cognitive impairment
 The Lewy body dementias
The Lewy body dementias
 Autoimmune antibody-associated encephalopathy and dementia syndromes
Autoimmune antibody-associated encephalopathy and dementia syndromes
 Sleep issues in dementia
Sleep issues in dementia
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


