Pseudotype envelope
Tropism and other features
References
Amphotropic MLV
Efficient transduction of hematopoietic progenitor cells, T lymphocytes
Reviewed in [60]
Aura virus
Transduction of monocyte derived dendritic cells (DCs), useful for genetic modification of DCs and DC-based immunotherapy
[130]
Baculovirus GP64
Transduction of glial cells; concentration via ultracentrifugation; nontoxic to host cells; inactivated by human complement
Reviewed in [60]
Ebola virus
Transduction reported for lung epithelial cells from the apical surface
[131]
Feline endogenous retrovirus (RD114)
Unmodified RD114GP: transduction of human mesenchymal stem cells; RD114-TR chimeric (transmembrane domain): transduction of human primary blood lymphocytes and CD34+ cells.
Feline leukemia virus
Transduction of human hematopoietic stem cells
[134]
Foamy virus
Efficient transduction of human neuroblastoma cell lines
[135]
Gibbon ape leukemia virus (GaLV)
Pseudotyping incompatibility with HIV-1. When cytoplasmic tail substituted with MLV transduction of primary human hematopoietic cells.
[136]
In baboons, transduction of marrow repopulating cells.
[137]
Influenza HA (hemagglutinin)
Transduction of respiratory epithelium (not effective)
Reviewed in [60]
Lymphocytic choriomeningitis virus (LCMV)
Efficient transduction of fibroblasts, epithelial cells, hematopoietic cell, hepatoma, neuroblastoma, glioma cell lines, neural stem cells
Measles virus
Generally transduction of lymphocytes, human monocyte-derived DCs (MDDCs)
Mokola virus
Retrogradely transported from periphery to CNS; retrograde and anterograde transport within the CNS; efficient transduction of human neuroblastoma cell line, rat astrocytoma cell line
Murine leukemia virus (MLV)
Transduction of human cord blood-derived CD34+ cells and clonogenic progenitors.
Transduction of preactivated T lymphocytes
Rabies virus
Distal targeting of neurons by retrogradely transport following intramuscular, intrastriatal, and intraspinal injection of RV-pseudotyped EIAV/HIV vectors, efficient neuronal transduction
Ross river virus (RRV)
RRV-pseudotyped FIV vectors: efficient transduction of hepatocytes and Kupffer cells in the liver upon systemic administration, neuroglial cells transduction (astrocytes and oligodendrocytes) upon brain injection
[147]
[43]
Sendai virus (SEV F and HN)
Transduction of apical surface airway epithelium. Human hepatocytes.
Sindbis virus
Can be used for cell specific targeting applications by insertion of cell-specific ligands; able to pseudotype oncoretroviruses and lentiviruses; efficient gene-targeting system based on antibody-mediated binding; efficient transduction of mouse liver and spleen cells upon intravenous injection
Venezuela equine infectious virus
Mostly neurons than glial
[151]
Vesicular stomatitis virus (VSV)
Broad cell tropism; preferentially transduces neurons when injected in the CNS
[53]
[152]
[59]
Concentration via ultracentrifugation
[131]
Not retrograde transport; cytotoxicity; inactivation by human serum
[145]
As previously discussed, several types of viral vectors are able to be retrogradely transported from the synapse, via the axon, to the cell body of neurons upon internalization at the axon terminal. These properties can be advantageous for precise and noninvasive remote gene delivery from accessible sites such as muscle or peripheral nerves to inaccessible areas of the CNS and might be applicable for treatment of a variety of neurodegenerative diseases. Remote access to the spinal cord via muscle injection can provide a relatively simple palliative treatment to spare key muscle groups such as those that control limb movement, swallowing and breathing to enhance the quality of and/or extend life. Direct intra-parenchymal spinal cord injection of vectors able to transport on the retrograde direction can also provide simultaneous gene delivery to both upper and lower motor neurons. As mentioned above, rabies-G pseudotyped EIAV vectors resulted in gene expression in the spinal cord upon intramuscular administration [62]. Nonetheless, each approach poses limitations and challenges for human application. The high affinity of some vectors in muscle, the architecture of nerves innervating the distal extremities, and more importantly the muscle mass, make intramuscular approach for treatment of motor neuron diseases one of the biggest challenges. Alternatively, injections into the peripheral nervous system have been attempted. Indeed Tanase et al. reported rabies-G EIAV gene expression in cervical spinal cord motor neurons in a mouse model upon brachial plexus injection [45]. However, the restricted access at synaptic terminals, which restricts both neuronal uptake and axonal transport, is a limiting factor for the application of this method in humans.
1.7 Cell Type-Specific Targeting
A desired gene therapy protocol would be to precisely deliver a gene of interest to specific cells in vivo using a targeted gene delivery vehicle for administration. As an alternative to pseudotyping with existing envelopes, various modifications have been employed to improve the targeting of vectors to specific cell types and regulate transgene expression.
Cell type-specific lentiviral vectors can be generated through surface engineering, by incorporating cell type–specific ligands or antibodies on a mutated viral envelope.
Several attempts have been made to alter the receptor-recognition attachment function in the envelope glycoprotein, without affecting membrane fusion [64]. Such modifications ablate the native tropism of the vector, so it is no longer able to recognize and bind to its cognate receptor, and redirect its tropism to the corresponding cellular receptors recognized by the ligand or antibody. Targeting systems available at the moment involve, among others, antibody specific binding methods based on (1) molecular events of receptor binding and endocytosis followed by fusogen-induced pH-dependent membrane fusion [65–67] or (2) active membrane fusion [68–70].
In 2006 Yang et al. have proposed an efficient targeting method. The method involves the incorporation of an antibody and a fusogenic protein as two distinct molecules on the lentiviral surface, separating the recognition from the fusion function by providing them as separate molecules. The fusogen is modified so it lacks the ability to bind to cognate receptors, but retains the ability to trigger pH-dependent membrane fusion, thus is binding deficient but fusion competent [65, 67, 71, 72]. The specificity of this LV is solely determined by the antibody chosen to recognize a specific antigen on the surface of the target cells. Recently these targeted LVs have been optimized by using single chain antibodies (scFV) genetically fused to either Sindbis virus or Measles virus (MV) hemagglutinin envelope [69, 73]. Modifications applied to optimize this method, aimed to overcome limitations such as receptor internalization and endosomal release [66], which are necessary when vectors are pseudotyped with certain envelopes.
An additional targeting method involves coating of the viral surface with polymers such as polyethylene glycol, poly-[N-(2-hydroxypropyl) methacrylamide], or biodegradable alginate microparticles [74–76]. All these methods are based on the same principle: ablating native tropism and retargeting through surface display of ligands (peptide, proteins, or antibodies). One disadvantage of the method is that the two-component nature of these bispecific molecules adds complications in vector production and in maintaining stable batch-to-batch homogeneity, as the number of plasmids required for production of these molecules targeted vectors increases (usually 5–6 plasmid co-transfection is required). Other approaches involve the use of a ligand protein or antibody as a bridge to attach the virus to specific cells [67, 71, 77, 78]. However, this approach requires endocytosis for the pH-dependent fusion in addition to the fact, that once the envelope protein is connected to the one end of the bridge molecule, it fuses inefficiently. Additionally to targeting through surface engineering, tissue specific promoters [79, 80] and or tissue-specific regulatory elements [81] have been used and might also optimize the targeting strategy restricting transduction to the tissue of interest. The use of mammalian promoters can result in long-term physiological levels of transgene expression [8]. Glial fibrillary acidic protein (GFAP), synapsin 1 [82–84], neuronal specific enolase (NSE) [85], tyrosine hydroxylase/neurofilament [86], and the prion promoters [87] are some examples of powerful neuronal promoters which have been used in adenoviral and lentiviral systems. Cytomegalovirus (CMV), platelet growth factor-β, and CMV/chicken β-actin promoters have also been tested in AAV and lentiviral vector systems for CNS targeting, where specificity achieved from each can vary between different CNS regions [84].
Alternatively, incorporation of drug-inducible systems (e.g., tetracycline, ecdysone, mifepristone [88, 89]) in lentiviral vectors can better control the levels and timing of gene expression [90]. Such lentiviral systems have been generated and used to mediate regulated gene delivery in Parkinson’s disease [90, 91]. Additionally, it is also possible to achieve cell type specific expression through microRNA (miR) regulation [92, 93]. When shRNA molecules are incorporated into a lentiviral vector, they are able to inhibit mRNA by RNA interference [94]. Moreover, control might be exerted by the inclusion of matrix scaffolding domains which place sequences in transcriptionally favored regions of the nucleus, and insulating elements which can protect promoters from the influences of vector and genomic sequences [95].
As described, transgene expression can be achieved using both integrating and non-integrating LVs. There is, however, a low risk of activation of proto-oncogenes or dedifferentiation following integration, as most transduced neural cells are terminally differentiated [4]. Concerns associated with random integration and insertional oncogenesis can be alleviated with the use of integration deficient lentiviral vectors (IDLV) that are deficient in integrase activity, the enzymatic activity required to catalyze integration into the host cell genome [96]. IDLV can be used for short-term expression of a gene of interest in dividing cells. As far as non-dividing target cells are concerned, specifically brain, liver, or retina, IDLV can induce relatively stable transgene expression [97–99]. Particularly, in short-term studies, it has been demonstrated that use of IDLV resulted in successful transduction of the rat striatum and retrograde transport to the dopaminergic neurons of the substantia nigra [99].
Moreover, IDLV can be used in conjunction with zinc-finger nuclease (ZFNs) hybrid technology to achieve site-specific gene editing/correction or addition at the targeted chromosomal loci [100] in which integration is believed to be safe, which could minimize or overcome the risks associated with random integration. Despite the fact that this technology is promising, requires further optimization in terms of both efficiency and safety. Nonetheless, the necessity for introduction of double-stranded DNA breaks (DSB) can be an issue as there is a potential of off-target effects where DSBs can be introduced in loci different from the desired one. This could result in accumulation of ZNFs to these sites and further contribute to ZFN cytotoxicity [101].
1.8 Lentiviral Vectors and Gene Therapy
Disorders of the nervous system still remain a major medical challenge, in matters of management and care. It is crucial to investigate and understand the disease mechanisms, identify therapeutic targets, and design and develop strategies for therapy. As already mentioned above, the ability to transfer genes to neuronal cells utilizing viral vectors and specifically lentiviral vectors, allows delivery of therapeutic genes to the nervous system. Neural architecture itself is promising for designing therapy. The morphology of each neuron and the interconnected networks they create, make the nervous system particularly appropriate for vector-mediated gene transfer; thus delivery at one site can lead to action at a distal site, if retrograde transport is possible [4]. Gene therapy approaches include delivery of trophic factors to protect neurons from damage, axogenic or regeneration molecules to restore neural function after injury, siRNA targeted to defective intracellular proteins and genes, or restoring missing proteins by gene replacement. Protocols of gene delivery for neurodegenerative disorders often requires invasive surgery and therefore repeat treatments are impractical; long-term, stable expression at therapeutic levels is essential. The therapeutic potential of viral vector gene therapy has been illustrated in many studies using animal models for human disease. The first use of lentiviral vectors for therapeutic gene expression (Bcl-xL) in the CNS was reported in 1998 [102]. Since then, effective long-term treatment has been reported in many animal models of neurological disorders, such as Parkinson’s disease (PD), Alzheimer’s disease, Huntington’s disease (HD), motor neuron diseases, LSD, and spinal cord injury (reviewed in [4, 43]).
LVs have also been efficiently used to develop transgenic disease models in rats and primates, which is important for behavioral studies. This technology allows over expression of the disease causing transgenes, which is essential for developing clear disease pathology and compared to standard transgenic techniques enables targeting of well-defined brain regions. Models of HD have been developed by using LVs overexpressing parts of the mutated huntingtin gene in the striatum of rats [103–105]. Indeed it was possible to induce pathological changes resembling those of HD. Similar to HD, lentiviral vectors have also been used to create novel animal models for PD by overexpressing α-synuclein in the dopaminergic cells of rats and primates [106–108]. On this front, therapeutic strategies have also been tested beyond the dopamine replacement and neurotrophic support approaches presented before [109]. Localized disease models can also be developed with vectors expressing siRNA using regulatable promoters or by applications of the Cre-lox recombination systems [110, 111].
The research applications of LVs presented above are promising for revealing information critical for our understanding of the nervous system in health and disease and for developing strategies to deliver therapy for neurological disorders. Table 2 summarizes the various gene therapy approaches for neurodegenerative diseases.
Table 2
Lentiviral vector-mediated approaches for neurodegenerative diseases
Gene therapy approach | Application | References |
|---|---|---|
Anti-apoptotic genes | Delivery of anti-apoptotic gene Bcl-XL for treating Alzheimer’s disease, protects neurons from neurotoxicity | [102] |
Gene Replacement | Lentiviral vectors expressing the normal SMN1 gene in models of spinal muscular atrophy | [19] |
Transplantation of cells, transduced ex vivo for tyrosine hydroxylase expression | [153] | |
[154] | ||
Knockdown of expression | Lentiviral vectors expressing siRNA targeted to mutant SOD1 gene in a model of amyotrophic lateral sclerosis | |
siRNA against axon growth inhibitors to promote axon regeneration in animal models of spinal cord/nerve injury | [157] | |
Knockout of mutant huntingtin with an siRNA in animal models of Huntington’s disease | [158] | |
Lentivirus-mediated siRNA targeted against BACE-1 to reduce amyloid production and behavioral deficits in mouse models of Alzheimer’s disease | [159] | |
Neurotrophic Factors | Localized lentivirus-mediated GDNF or BDNF delivery protects the surviving neurons of the nigrostriatal dopaminergic pathway from further degeneration in rodent models of Parkinson’s disease. | |
Lentiviral vectors expressing VEGF for treating animal models of motor neuron diseases | [62] | |
Lentiviral vectors expressing CNTF and BDNF for treating animal model of Huntington’s disease | [166] | |
Lentiviral vectors expressing GDNF protected facial motor neurons from axotomy-induced cell death | [167] | |
Engrafted fibroblasts retrovirally engineered to secrete NGD prevented degeneration of the basal forebrain cholinergic neurons in rat and primate models of Alzheimer’s disease | [168] | |
Ex vivo transduction of Schwann cells and olfactory ensheathing cells to express neurotrophin for the promotion of axonal regeneration in spinal cord injury models. | [169] | |
Tet-regulated lentiviral vector expressing NGF to rescue cholinergic neurons in the fimbria-formix of rats models of Alzheimer’s disease | [170] |
2 Materials
The selection of basic materials and proper preparation of reagents required for lentiviral production is critical for success. Indeed for many of the materials it is recommended that these specific vendors and catalog numbers be utilized. A complete list of stock reagents needed, including catalog number and vendor name, as well as equipment required is provided in Tables 3 and 4. Cell culture conditions for all procedures in this chapter utilize a standard tissue culture incubator, with 5 % CO2, humidified environment at 37 °C. Lentiviral vector production should be carried out in a Biosafety Level 2 (BSL2) approved facility. We provide recipes for all media and solutions described in the methods in Sect. 3.
Table 3
Components and concentrations of culture media, buffers, and other solutions
Volume | Components | Final concentration |
|---|---|---|
Complete DMEM media | ||
440 ml | DMEM (SIGMA, # D6546) | |
50 ml | Newborn Calf Serum (Heat Inactivated) (SIGMA #N4762-500ML) | 10 % |
5 ml | Pen/Strep (100×, SIGMA # P4333) | 1× |
5 ml | l-glutamine (100×, SIGMA # G7513) | 1× |
Induction media | ||
480 ml | DMEM (Sigma, UK # D6546) | |
10 ml | Newborn Calf Serum (Heat Inactivated) (SIGMA #N4762-500ML) | 2 % |
5 ml | Pen/Strep (100×, SIGMA # P4333) | 1× |
5 ml | l-glutamine (100×, SIGMA # G7513) | 1× |
Induction solution (always prepared fresh) | ||
5 ml | Sodium Butyrate 1 M solution | 10 mM |
495 ml | Induction Media | |
Polybrene 100× | ||
40 mg | Hexadimethrine bromide (Polybrene) (Sigma, #107689) | 40 mg |
50 ml | DULBECCO’S PHOSPHATE BUFFERED SALINE (PBS) (SIGMA, # D8537) | 50 ml |
Sodium butyrate (NaBut 1 M) | ||
0.55 g | Sodium Butyrate (ALDRICH, # 303410) | 1 M |
5 ml | Cell culture grade H2O (SIGMA, # W3500) | |
TSSM formulation buffer | ||
0.12 g | Tromethamine (SIGMA, # T6687) | 20 mM |
0.29 g | NaCl (SIGMA, # S7653) | 100 mM |
0.5 g | Sucrose (SIGMA, # S9378) | 10 mg/ml |
5 ml | Mannitol (SIGMA, # M9546) | 10 mg/ml |
Up to 50 ml | Cell culture grade H2O (SIGMA # W3500) | |
TVM1 media (pH 7.10) | ||
100 ml | Cell culture grade H2O (SIGMA # W3500) | |
1.38 g | DMEM-H powder (SIGMA D5648-SL) | |
2.5 ml | 1 M HEPES (SIGMA H3537) | |
TVM2 media (pH 7.90) | ||
326.8 ml | Cell culture grade H2O (SIGMA # W3500) | |
5.52 g | DMEM-H powder (SIGMA D5648-SL) | |
10 ml | 1 M HEPES (SIGMA H3537) | |
40 ml | Newborn Calf Serum (SIGMA) | 10 % |
19.72 ml | 7.5 % sodium bicarbonate solution (SIGMA, S8761) | |
4 ml | 10× Pen-Strep (Penicillin 10,000 U/ml; Streptomycin 10,000 μg/ml, SIGMA) | 1 % |
20 % Sucrose solution | ||
80 ml | Cell culture grade H2O (SIGMA # W3500) | |
20 g | Sucrose powder (SIGMA # 84097) | 20 % |
2 ml | 5 M NaCl (SIGMA, # S5886) | |
2 ml | 1 M HEPES solution pH 7.4 (GIBCO, #15630-049) | |
200 μl | 0.5 M EDTA solution | |
4 % Paraformaldehyde | ||
4 g | Paraformaldehyde powder (SIGMA # P6148) | 4 % |
100 μl | 10 M NaOH | |
10 ml | PBS (SIGMA, # D8537) | |
85 ml | Cell culture grade H2O (SIGMA # W3500) | |
1 M CaCl2 | ||
11.1 g | CaCl2 powder (SIGMA # 22350-6) | 1 M |
100 ml | Cell culture grade H2O (SIGMA # W3500) | |
Table 4
List of reagents and consumables
Reagents and consumables | Sources and catalog numbers |
|---|---|
Ampicillin sodium salt | SIGMA, A9518-5G |
Cryotube 1.0 ml | Nunc, 366656 |
DAPI | SIGMA |
1 kb DNA ladder | NEB, #N3232 |
EndoFree plasmid mega kit [5] | QIAGEN, 12381 |
50 ml Falcon tubes | VWR, 525-0156 |
HEK 293T cell line | ATCC (CRL-11268™) [HEK293T/17] Rockefeller Univ. |
Lenti-X qRT-PCR titration kit | Clontech, Cat. No. 631235 |
Newborn calf serum (heat inactivated) | SIGMA, #N4762-500ML |
OCT | Surgipath FSC22, Leica Microsystems, Wetzlar, Germany |
Opti-MEM® I reduced serum medium, no phenol red | Gibco, Life Sciences, # 11058021 |
Phosphate buffer saline (10×) | SIGMA, #D8537 |
ProLong gold antifade reagent | Invitrogen, P36930 |
Poly-l-lysine coated slides | VWR, 631-0107 |
Polypropylene ultracentrifugation tube | Beckman, 337986 |
Retro-Tek HIV-1 p24 antigen ELISA kit | Zeptometrix, 0801200 |
Screw top centrifuge tubes | Nalgene |
Triton X-100 | SIGMA, X100 |
Trypsin-EDTA | SIGMA, #T4174 |
0.20 μm Filter (500 ml capacity) | Thermo Scientific, 162-0020 |
0.45 μm Filter (500 ml capacity) | Thermo Scientific, 162-0045 |
0.20 μm Syringe filter | Nalgene, 190-2520 |
0.45 μm Syringe filter | Nalgene, 190-2545 |
T175 cm2 flasks | Nunc, 159910 |
T75 cm2 flasks | Nunc, 156499 |
12-Well plate | Nunc, 150628 |
24-Well plate | Nunc, 142475 |
150 × 25 mm Tissue culture dish | BD Falcon, 353025 |
UV half area plate, 96 well | Corning, 3679 |
Vybrant multicolor cell-labeling kit (DiO, Dil, DID solutions) | Life Sciences, #V22889 |
pRRLsin–cPPT–CMV-eGFP–WPRE | Transgene cassette contains a self-inactivating 3′LTR. eGFP |
Reporter gene driven by CMV promoter, also codes for ampicillin resistance | |
pMD2–LgpRRE | Structural protein is within a pMD2 plasmid backbone carrying an ampicillin resistance gene and driven by a Cytomegalovirus (CMV) promoter |
pRSV–Rev | Accessory protein situated within plasmid carrying ampicillin resistance gene and driven by the RSV U3 promoter |
pMD2–VSVg | VSV envelope glycoprotein driven by a CMV promoter and encodes for ampicillin resistance gene. |
3 Methods
3.1 Preparation and Quantification of Plasmids Required for Viral Production
3.1.1 Plasmid DNA Extraction/Purification
We recommend purifying all plasmids for lentiviral production using EndoFree plasmid purification kits (Qiagen)—and following the standard protocol in the Mega kit handbook. For details of the reagents and procedures used refer to handbook or company website (www.qiagen.com/) (see Note 1).
1.
Prepare a starter culture for each plasmid in 50 ml Falcon tubes by the addition of 5 ml LB, 5 μl of ampicillin stock solution (50 mg/ml) to a final concentration 50 μg/ml, and inoculate with a single colony from a transformation plate, using an inoculating loop.
2.
Incubate in a shaking incubator (250 rpm) for 6–8 h at 37 °C.
3.
When the culture medium becomes turbid due to bacterial growth, take 1 ml of the starter culture and inoculate 500 ml of LB with 50 μg/ml ampicillin.
4.
Incubate overnight (12–16 h) in a shaking incubator (300 rpm) at 37 °C.
5.
Next day, transfer the bacteria to 500 ml screw-top centrifuge tubes (Nalgene) and harvest the bacteria by centrifugation at 6,000 × g for 15 min at 4 °C.
6.
Continue all subsequent procedures of plasmid purification using Qiagen EndoFree Mega kits. Following purification, resuspend the DNA pellet in 400 μl TE buffer and store at −20 °C.
3.1.2 Determination of DNA Quality and Concentration by Spectrophotometry
Calculation is made based on the ratio of absorbance at 260 nm (DNA) and 280 nm (proteins) analyzed by spectrophotometry (Promega GloMax-Multi Detection System).
1.
Dilute DNA samples of each purified plasmid 1:300 and 1:600 in Tris-EDTA (TE) Buffer (average DNA concentration of ~0.05 μg/μl). Use TE buffer also as a reference value.
3.
Load the plasmid samples into a 96 well Corning 3679 plate and measure the A260/A280 ratio for each sample.
4.
Use the following formula to calculate DNA concentration:
Concentration of dsDNA (mg/ml) = A260nm/(dsDNA extinction coefficient at 260 nm × path length) × dilution.
Where extinction coefficient is 20 (mg/ml × cm) and path length of each well is calculated as 0.6.
3.1.3 Supercoiled DNA gel Quantification by Densitometry
The concentrations calculated from the measurements of spectrophotometer are of total DNA, and it is advisable to calculate the concentration of supercoiled DNA as it has been shown that supercoiled DNA is a more accurate representation of what becomes transfected. Supercoiled DNA can enter cells more efficiently than relaxed plasmids during transfection (calcium phosphate) and furthermore, surplus DNA can be toxic to the cells, highlighting the requirement for quantification of supercoiled DNA in addition to that obtained from spectrophotometry. This is carried out by a gel based quantification method as follows:
1.
Dilute plasmid DNA samples. Dilution is calculated from the spectrophotometric reading (<1 mg/ml, 1:10 dilution; between 1 and 4 mg/ml, 1:100 dilution; and >4 mg/ml 1:200 dilution).
2.
Analyze samples by gel electrophoresis by running increasing concentrations of each sample alongside equally increasing concentrations of a 1 kb DNA ladder (500 μg/ml).
3.
Prepare sample and ladder concentrations as presented in Fig. 1a.
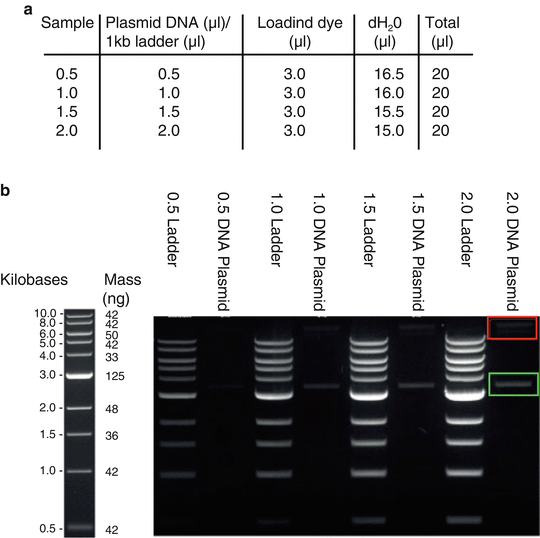
Fig. 1
Gel quantification of supercoiled DNA. (a) Schematic presentation of DNA sample dilutions. Load 20 μl of each plasmid and ladder sample dilutions on 1 % agarose gel and allow running at 80 V. (b) Example of gel image obtained from quantification of pMD-RVGCVS24-B2c plasmid. Left: NEB 1 kb ladder; Right: Increasing concentrations of ladder and plasmid (0.5–2.0 μl) samples were used to generate an average. Supercoiled DNA (green box) is aligned to the closest ladder band (here 3 kb) and DNA concentration is quantified by densitometry using equivalent mass (ng) of the selected ladder band (here 125 ng). Non-supercoiled/relaxed DNA (red box) is omitted
4.
Load both ladder and DNA samples to a 1 % agarose gel containing 10 μl of (10 mg/ml) ethidium bromide as presented in Fig. 1b.
5.
Run gel at 90 V for 40–45 min until ladder unfolds properly.
6.
The band intensity of each DNA sample was analyzed by Silicon software to work out its supercoiled DNA concentration by comparing to the band intensity of standard 1 kb DNA ladder.
A representative gel image of B2c-RVG plasmid is presented in Fig. 1b. In the present example, the ratio of supercoiled DNA over total DNA is 3:1.
3.1.4 Restriction Enzyme Analysis of DNA Samples
We recommend you always perform suitable restriction enzyme diagnostic digests for all plasmids that will be used for viral production to confirm their identities and visually evaluate the quality of the DNA samples.
3.2 Lentivirus Production
For biosafety reasons, lentiviral vector production for gene therapy is designed to be replication deficient by separating cis– (important in viral genome transfer) and trans-acting sequences (important for viral packaging) to prevent production of replication competent lentivirus (RCL). Production of lentiviral vectors is routinely achieved by transient co-transfection of human embryonic kidney (HEK 293T) cells using high concentrations of three or four different plasmids. These are: (1) the lentiviral expression plasmid (transfer plasmid) which encodes the transgene of interest flanked by all cis-acting viral sequences required for packaging of RNA genome, (2) one or two packaging plasmids which encode the viral structural and functional proteins, and (3) the pseudotyping (envelope) plasmid which encodes for the glycoprotein responsible for receptor recognition. The most commonly used glycoprotein is VSV-G [13, 112–114]. Viruses are obtained by collecting supernatants from the transfected packaging cells. Typical titers of lentiviral vectors pseudotyped with VSV-G range from 106 to 107 transducing units per milliliter. Increased viral titers can be achieved by physical concentration by ultracentrifugation. Lentiviral vectors pseudotyped with VSV-G have proven to be stable and able to withstand concentration by ultracentrifugation without this having an effect to the titer [115]. Highly concentrated viral preparations are required for in vivo applications. Viral containing supernatants are concentrated and stored at −80 °C for future use. It is preferable that viral preparations are used fresh due to concerns of loss of viral titer resulting from freeze–thaw cycle. The production and complete titration protocols described below can be completed in 12 days (see Fig. 2). All plasmids required for virus production are listed in Table 4.
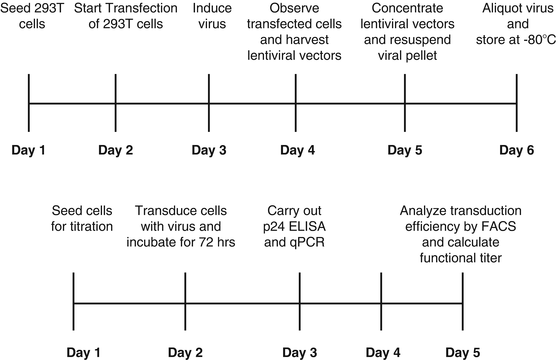
Fig. 2
Timeline of lentiviral vector production and titration
3.2.1 Propagation, Passaging, and Preservation of Packaging Cell Line HEK 293T
Successful lentiviral vector production resulting in high titers is tightly connected to the state of HEK 293T cells used. It is critical to use healthy cells at low passage numbers. Thus, a large stock of HEK 293T cells at a lower passage number should be kept for future production.
Thawing the HEK 293T Cells
1.
Rapidly transfer the cryovial containing the HEK 293T from the liquid nitrogen storage into a 37 °C water bath.
2.
Thaw the cells by gentle agitation until most, but not all, of the contents is thawed.
3.
Remove the vial from the water bath and let the remaining cells thaw.
4.
Rinse the vial with 75 % ethanol before transferring it into the tissue culture hood.
5.
Transfer the thawed cell suspension into a T175 flask containing pre-warmed complete DMEM medium.
6.
Transfer the flask into the incubator and incubate for 4 h until the majority of cells attach to the bottom of the flask.
7.
Four hours later replace the medium with pre-warmed fresh medium to the cells.
8.
Next day examine the cells and check for cell death. Replace the medium with fresh one.
9.
Passage the cells when they are 75–80 % confluent.
10.
Change the medium every 2 days.
Passaging HEK293T Cells
1.
Grow cells at 37 °C in an incubator.
2.
Aspirate the complete DMEM medium from a T175 tissue culture flask when the cells have reached 75–80 % confluency. Do not allow the cells to reach >90 % confluency.
3.
Wash once with 25 ml of PBS and aspirate.
4.
Add 2.5 ml of 1× trypsin-EDTA and incubate at 37 °C for 2–3 min.
5.
Add 7.5 ml of complete DMEM medium and gently pipette up and down to detach the cells, creating a single-cell suspension (see Note 3).
6.
When passaging the cells for the first time after thawing, it is preferable to propagate the cells at two different ratios (1:5 and 1:10) and follow the growth and replication rate.
8.
Three days prior to transfection prepare suitable number of T175 flasks with HEK 293T cells to achieve suitable number of cells required for viral production.
It is usually preferable to allow cells to adjust to the medium and the propagating conditions for 1–1.5 weeks (two passages) before using them for viral production. It is of high importance to completely renew the medium the day prior to plating cells for viral production. A timeline regarding lentiviral production and titration is presented in Fig. 2.
3.2.2 Transfecting HEK 293T Cells
1.
Twenty-four hours prior to transfection (day 1), plate twelve 150 mm cell culture dishes with 1.4 × 107 HEK 293T cells at a density of ~7.6 × 104 cells cm−2 in 16 ml complete DMEM and incubate overnight in a standard tissue culture incubator.
2.
The next day (day 2), 3 h prior to transfection, make and warm to room temperature fresh TVM1 and TVM2 media. The reagents required are listed in Table 3 (see Note 4).
Procedure
TVM1: In a T75 flask, make up 100 ml serum-free DMEM-H without sodium bicarbonate by the addition of 1.38 g DMEM-H powder to 100 ml sterile tissue culture water. Add 2.5 ml 1 M HEPES and then transfer the flask to the magnetic stirrer. Stir until all powder has dissolved, then adjust to pH 7.10 by addition of 1 M NaOH. Sterilize by filtration using a 0.22 μm syringe filter and store at 4 °C.
TVM2: In a clean autoclaved 500 ml Duran bottle or T175 flask, make up 400 ml complete DMEM-H by the addition of 5.52 g DMEM-H powder to 326.8 ml sterile tissue culture water while swirling gently. Add 19.72 ml 7.5 % sodium bicarbonate solution. Add 10 % newborn calf serum (NCS), 1× Pen-Strep, and 10 ml 1 M HEPES. Adjust to pH 7.9 by addition of 1 M NaOH. Sterilize by filtration using a 0.22 μm filter with 1 l capacity and store at 4 °C (see Note 4).
3.
Immediately before transfection prepare four 50 ml Falcon tubes and add to each 45 μg of lentiviral vector plasmid pRRL-sin-cppt-CMV-GFP-WPRE, together with 45 μg of pMD2-LgpRRE and 15 μg of pRSV-Rev packaging plasmids and 15.3 μg of envelope plasmid DNA (This is a typical four plasmid co-transfection. In other protocols where six plasmids are required for lentiviral production the quantity of plasmids can be adjusted [65]).
4.
Make each tube up to a final volume of 8 ml with TVM1.
5.
Add 410 μl 1 M CaCl2 in two stages; first drop in 5–6 drops while mixing by flicking or slightly vortexing and then add the remaining CaCl2 quickly without mixing.
6.
Incubate the reaction mixtures at room temperature for 10 min.
7.
During the last 4 min of incubation examine the HEK 293T in the dishes and carefully remove the complete DMEM medium.
8.
Add 32.8 ml of TVM2 medium to each Falcon tube and mix by pipetting up and down.
9.
Immediately after mixing add 13.5 ml of the DNA/TVM1/TVM2 transfection mixture to each dish, taking care not to dislodge the cells.
10.
Return the dishes to the incubator in stacks of four (usually no more than six).
11.
Sixteen hours post-transfection (day 3) examine the cells under a microscope. If a reporter gene encoding a fluorescent protein, such as green fluorescent protein (GFP), is used under the control of a constitutive promoter (such as CMV), examine the cells under a fluorescence microscope for reporter gene expression. More than 70 % of cells should fluoresce green. Proceed to viral production if 60–70 % of producer cells express the reporter gene. If this is not the case, stop production and start again.
12.
13.
 Gene Therapy Approaches to Promoting Axonal Regeneration After Spinal Cord Injury
Gene Therapy Approaches to Promoting Axonal Regeneration After Spinal Cord Injury
 Gene Therapy Approaches Using Reproducible and Fully Penetrant Lentivirus-Mediated Endogenous Glioma Models
Gene Therapy Approaches Using Reproducible and Fully Penetrant Lentivirus-Mediated Endogenous Glioma Models
 Gene Therapy for Huntington’s Disease
Gene Therapy for Huntington’s Disease
 Gene Therapy for Chronic Pain: How to Manipulate and Unravel Pain Control Circuits from the Brain?
Gene Therapy for Chronic Pain: How to Manipulate and Unravel Pain Control Circuits from the Brain?
 Adeno-Associated Vectors for Gene Delivery to the Nervous System
Adeno-Associated Vectors for Gene Delivery to the Nervous System
 Gene Therapy for Parkinson’s Disease: AAV5-Mediated Delivery of Glial Cell Line-Derived Neurotrophic Factor (GDNF)
Gene Therapy for Parkinson’s Disease: AAV5-Mediated Delivery of Glial Cell Line-Derived Neurotrophic Factor (GDNF)




Thirty-six hours later (day 4) examine the cells under the microscope. Cells should be fused and multinucleated. This morphological change is expected and does not affect the production of lentiviral vectors. Most cells should still be attached and around 90–95 % should fluoresce green. To harvest the viral supernatants, collect the supernatant from the 12 dishes into four 50 ml Falcon tubes and centrifuge for 5 min at 750 rpm to remove viral and cell debris. Proceed with filtration through a 0.45 μm pore-sized filter (see Note 6).
Related posts:
Gene Therapy Approaches to Promoting Axonal Regeneration After Spinal Cord Injury
Gene Therapy Approaches Using Reproducible and Fully Penetrant Lentivirus-Mediated Endogenous Glioma Models
Gene Therapy for Huntington’s Disease
Gene Therapy for Chronic Pain: How to Manipulate and Unravel Pain Control Circuits from the Brain?
Adeno-Associated Vectors for Gene Delivery to the Nervous System
Gene Therapy for Parkinson’s Disease: AAV5-Mediated Delivery of Glial Cell Line-Derived Neurotrophic Factor (GDNF)
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
