Fig. 14.1
Cerebral form of X-linked adrenoleukodystrophy, c-ALD. MRI axial T2-weighted images (a, b) show marked bilateral symmetrical hyperintensity in the splenium of the corpus callosum and in the adjacent white matter. Post-contrast image (c) shows symmetrical linear peripheral contrast enhancement in the corresponding areas (arrows).
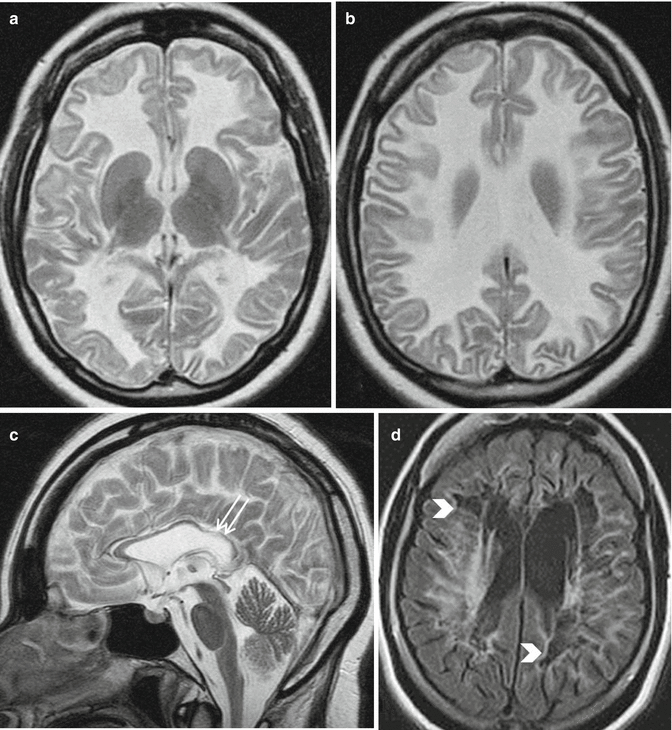
Fig. 14.2
Vanishing white matter. Axial and sagittal T2-weighted images (a–c) show extensive cerebral white matter abnormalities with involvement of “U” fibers; the corpus callosum is very thin and the inner rim of the corpus callosum is affected (c, arrows). Axial FLAIR image in an another patient (d) reveals almost complete disappearance of white matter with cystic degeneration (arrowheads)
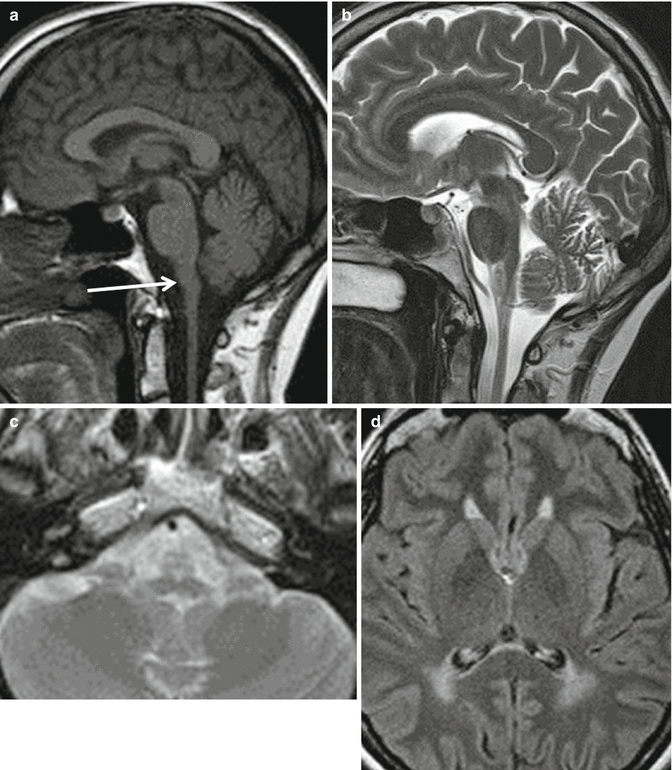
Fig. 14.3
Adult onset AxD. T1 and T2-weighted midline sagittal section (a, b) show atrophy and signal abnormalities in the medulla oblongata (arrow). Axial T2-weighted section (c) demonstrates signal hyperintensities in the atrophic anterior part of the medulla oblongata. Axial FLAIR image (d) shows increased signal intensity in the periventricular white matter, more extensive in the posterior regions
Table 14.1
Adult-onset leukodystrophies
MIM # | Mode of inheritance | Genes | Age at onset | Diagnostic tests, in addition to gene tests | Treatment available | |
|---|---|---|---|---|---|---|
Adrenoleukodystrophy/adrenomyeloneuropathy | 300100 | X-linked | ABCD1 | C-A | MRI, blood VLCFA | Yes |
Metachromatic leukodystrophy | 250100 | AR | ARSA | C-A | MRI, arylsulfatase A enzyme activity | Yes |
Krabbe disease | 245200 | AR | GALC | C-A | MRI, beta-gactocerebrosidase enzyme activity | Yes |
Adult polyglucosan body disease | 263570 | AR | GBE1 | A | MRI, nerve biopsy | No |
Autosomal dominant adult-onset leukodystrophy | 169500 | AD | LMNB1 | A | MRI | No |
Hereditary diffuse leukoencephalopathy with spheroids | 221820 | AD | CSF1R | A | MRI | No |
Nasu-Hakola disease | 221770 | AR | TREM2, TYROBP | A | MRI, skeletal X-rays | No |
Leukodystrophy with vanishing white matter | 603896 | AR | EIF2B1-5 | C-A | MRI | No |
Alexander disease | 203450 | AD | GFAP | C-A | MRI | No |
LBSL | 611105 | AR | DARS2 | C-A | MRI | No |
Pelizaeus-Merzbacher disease | 312080 | X-linked | PLP1 | C(−A) | MRI | No |
Pelizaeus-Merzbacher-like disease 1 | 608804 | AR | GJC2 | C-A | MRI | No |
Leukoencephalopathy with ataxia | 615651 | AR | CLCN2 | C-A | MRI | No |
There are few studies addressing the prognosis of adult-onset leukodystrophies, mainly because of their rarity. In general, the prognosis of the leukodystrophies presenting in adulthood seems to be better than for those with onset in infancy or childhood. Notwithstanding, their natural history is commonly characterized by relentless progression, and no disease-modifying therapy has been proved to be effective for the majority of them. Hematopoietic stem cell transplantation (HSCT) with or without gene therapy, however, is a promising treatment for some of them, and supportive care has improved the quality of life and prolonged the survival of most of the affected subjects.
14.2 Adrenoleukodystrophy (X-ALD)
Definition and epidemiology
Adrenoleukodystrophy (X-ALD) is an X-linked metabolic disease caused by mutations in the ABCD1 gene, and is the most common inherited leukoencephalopathy, with an estimated prevalence of hemizygotes (i.e., males) plus heterozygotes (i.e., females) of 1:16,800 in the USA.
Clinical features
The penetrance in males seems to be approximately 90 % [4], whereas X-ALD female carriers can develop neurological symptoms in 20–50 % of cases, usually in the fourth or fifth decade of life [5, 6]. In males, X-ALD encompasses a wide spectrum of forms with varying severity, ranging from the most severe cerebral form (c-ALD) – characterized by behavioral and cognitive decline as predominant manifestations – to the less severe adrenomyeloneuropathy (AMN) – characterized by spastic paraplegia, lower limb sensory disturbances, sphincter dysfunction and impotence as predominant manifestations – or even to adrenal failure-only phenotype. There is no genotype-phenotype correlation, and the same mutation can lead to different forms even within the same family.
Therapy
Lorenzo’s oil is not useful for c-ALD, whereas its efficacy for adrenomyeloneuropathy (AMN) remains uncertain because of a lack of randomized placebo-controlled studies in adulthood [7]. Heterologous hematopoietic stem cell transplantation (HSCT) from HLA-matched donor and autologous HSCT with gene therapy (i.e., lentiviral hematopoietic stem cell gene therapy) can be efficacious therapies for preventing the development of c-ALD in children [8], but AMN can develop later in life [9]. Notably, hematopoietic stem cell transplantation (HSCT) seems to be successful as a disease-modifying therapy also in subjects with AMN who develop cerebral demyelination, if they are treated as soon as cerebral demyelination occurs.
Prognosis
AMN is by far the most common phenotype among the adult-onset forms of X-ALD, while c-ALD or Addison-only phenotype rarely has onset or persists in adulthood. Indeed, c-ALD most commonly presents at the age of 7 ± 2 years and usually leads the patient to a vegetative state or death in 3 years or so. Signs of adrenocortical failure most commonly start between 5 and 10 years of age, but the majority of patients with adrenocortical failure – who can survive thanks to hormone replacement therapy – will develop AMN later in life [7]. The mean age at onset of AMN is 28 ± 9 years [7], but about 70 % of patients already have adrenocortical failure when the first neurological symptoms start. About 55 % of AMN patients do not develop cerebral demyelination at all (“pure” AMN), while the remaining 45 % can show cerebral lobar white matter involvement (adrenoleukomyeloneuropathy [ALMN]) on brain MRI, which may become symptomatic and variably progressive. In particular, over a period of 9.5 (±5.5) years, about 20 % of subjects with “pure” AMN can unpredictably develop severe cerebral demyelination, leading to total disability and death after a survival period of approximately 2 years (2.3 ± 1.9 years, median 1.6 years, range 0.5–7.6 years) [10]. A recent study, however, has concluded that cerebral demyelination can be higher than previously reported, given that 63 % of subjects with AMN developed brain involvement over a period of 10 (±6.9) years after the onset of myelo(neuro)pathy and died after 3.4 ± 2.9 years [11]. In “pure” AMN, walking disturbances (spastic gait) seem to be followed by sensory disturbances in the legs, then by bladder dysfunction (i.e., urgency, urinary incontinence, and urinary retention), and eventually by fecal urgency and difficulty to defecate, after a mean period of 5, 6, and 12 years, respectively [10]. Overall, the progression of the handicap is gradual, with a mean of the modified Rankin score of 1.7 (±0.9) and 2.9 (±1.1) after 9.1 (±8.2) and 16.2 (±8.9) years from disease onset, respectively. In particular, AMN patients seem to need assistance to walk and they can be even wheelchair-bound about 16 years after the first neurological symptoms [10].
In their 40s, around 50 % of X-ALD female carriers, and probably more when their age increases to ≥60 years, can develop mild myeloneuropathy with impaired vibration sense and lower limb hyper-reflexia and pain, while around 15 % of cases can develop an overt spastic paraparesis with bladder dysfunctions and fecal incontinence, resembling the male AMN form [5, 6]. As a rule, however, for female carriers life expectancy remains normal, with the exception of ultra-rare cases suffering from cerebral involvement resembling the male c-ALD form.
14.3 Adult-Onset Metachromatic Leukodystrophy (MLD)
Definition
Metachromatic Leukodystrophy (MLD) is a metabolic disease caused by recessive mutations in the gene arylsulfatase A (ARSA).
Epidemiology
MLD incidence is reported to be approximately 1 in 100,000 births, with the adult form (onset after age 16) affecting 20 % of individuals with the disease (2 cases per million per year).
Clinical features
Adult-onset (AO) MLD seems to be caused by mutations encoding ARSA with residual enzymatic activity (so-called R-alleles) and is characterized at onset by behavioral and cognitive decline, which may lead to the erroneous diagnosis of schizophrenia, or by walking difficulties caused by spastic paraparesis or cerebellar dysfunction. The onset is usually in the twenties, but it can also occur at age 60 or later [1, 12].
Prognosis
The evolution is slowly progressive, but periods of relative stability may be seen (http://ghr.nlm.nih.gov/condition/metachromatic-leukodystrophy). Although some patients with isolated behavioral and cognitive decline at onset do not develop any neurological symptoms despite a disease duration of more than 20 years, most of them develop spastic or ataxic motor impairment over a median period of 7 years after disease onset (range 2–14 years) [12]. Similarly, patients with walking difficulties at onset usually develop neuropsychiatric symptoms over a period of months or years. Hence, the two different types of MLD patients at onset become clinically similar in advanced disease. Optic atrophy, signs of bulbar dysfunction such as dysphagia, and epileptic seizures may appear during disease evolution, and in the terminal stages, the patients have a severe dementia and eventually reach a vegetative state lying in a decorticate or decerebrate posture [1]. Subjects with AO-MLD may survive from 1 to 20–30 years after disease onset, with a mean of about 7 years [12]. Allogeneic hematopoietic stem cell transplantation (HSCT) seems to be able to halt the progression of late MLD [13], whereas lentiviral hematopoietic stem cell gene therapy seems to be effective in preventing the onset of the disease in presymptomatic subjects, thus opening a new era in the therapy of MLD [14].
14.4 Adult-Onset Krabbe Disease
Definition
Krabbe disease is a rare autosomal recessive metabolic leukodystrophy caused by mutations in the galactosylceramidase (GALC) gene.
Epidemiology
The incidence of Krabbe disease is approximately 1 in 100,000 births.
Clinical features
The onset is usually in the first 6 months of life (early infantile form), and the prognosis is very poor, because the course is rapidly progressive with death within approximately 1 year after the onset. In contrast, Krabbe disease with onset in childhood can persist into adulthood [15], and HSCT can be effective in arresting the progression of the disease [16]. The adult-onset form (≥16 years) is very rare (<10 % of the individuals with the disease), and nearly constantly presents with progressive walking difficulties due to spastic paraparesis or (asymmetric) lower limb weakness. The mean age at onset in 25 patients with adult-onset Krabbe disease was 35 years, ranging from 16 to 66 years [15].
Prognosis
Disease evolution is slowly progressive, but rapid decline within a few months and periods of relative stability have been reported. Walking difficulty remains the main symptom, with other neurological symptoms, such as dysarthria, cerebellar ataxia, pes cavus, altered vibration sense, optic atrophy/pallor, tongue atrophy/fasciculation, urinary dysfunction, and/or cognitive decline developing in a minority of subjects. The mean time to the wheelchair-bound state seems to be approximately 15 years, and severe dementia is rare [15]. Unlike the infantile form, life expectancy in adult-onset patients maybe normal (http://ghr.nlm.nih.gov/condition/krabbe-disease), although respiratory failure and aspiration pneumonia from severe dysphagia can lead to premature death [15].
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


