SECTION IV LOCALISED NEUROLOGICAL DISEASE AND ITS MANAGEMENT C. PERIPHERAL NERVE AND MUSCLE
THE POLYNEUROPATHIES – FUNCTIONAL ANATOMY
STRUCTURE OF THE NERVE CELL AND AXON
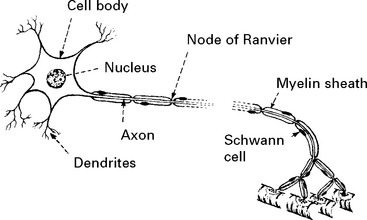
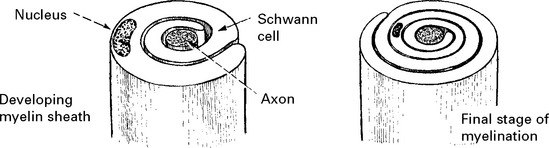
All axons have a cellular sheath – Schwann cell – but not all axons are myelinated.
SPINAL PERIPHERAL NERVOUS SYSTEM
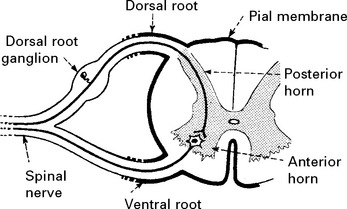
Sensation can be divided into:
These different forms of sensation are carried from the periphery by axons with specific characteristics. The central connections and pathways vary also (see page 200).

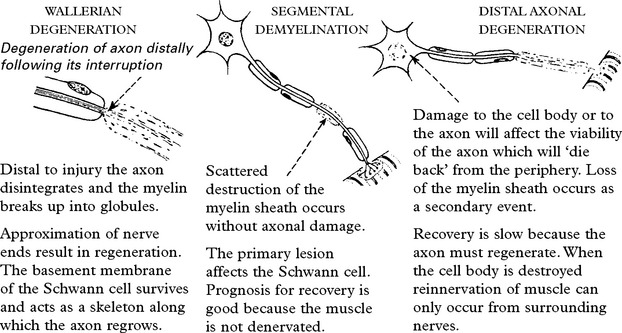
THE POLYNEUROPATHIES – SYMPTOMS
Sensory
Negative phenomena – loss of sensation.
Disease of large myelinated fibres produces loss of touch and joint position perception.
Disease of small unmyelinated fibres produces painful positive phenomena:
International Association for the Study of Pain (IASP) definitions has clarified the following.
| absent sensitivity to a painful stimulus | |
| increased sensitivity to a painful stimulus | |
| reduced sensitivity to painful stimulus | |
| increased sensitivity to any stimulus | |
| reduced sensitivity to any stimulus | |
| increased sensitivity with increasing threshold to repetitive stimulation | |
| pain provoked by a non-painful stimulus |
THE POLYNEUROPATHIES – SIGNS
SENSORY EXAMINATION


Examination of gait is important; with joint position impairment, sensory ataxia is evident. Romberg’s test is positive (see page 28). Neuropathic burns/ulcers or joints may be present.
The AXON REFLEX can be used to ‘place’ lesions in the sensory pathway.
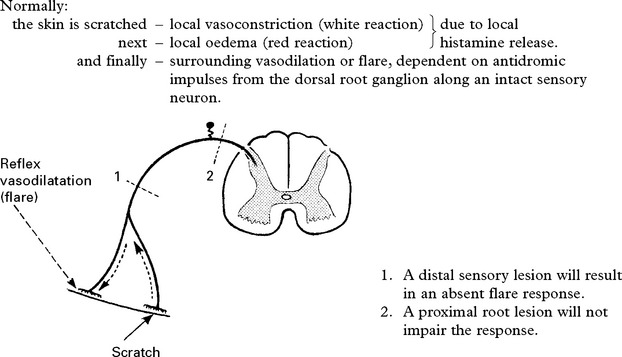
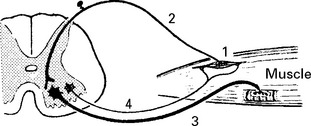
THE POLYNEUROPATHIES – CLASSIFICATION
There are several approaches to classification:
The following table based primarily on mode of onset is for reference. Certain neuropathies will be dealt with separately (see pages 439–444).
| CAUSE | FUNCTIONAL DISTURBANCE | PATHOLOGY |
|---|---|---|
| ACUTE: days to 4 weeks | ||
| Inflammatory (Guillain Barré syndrome) | Predominantly motor | Demyelinative with perivascular lymphocytic infiltration |
| Distal or proximal | ||
| Autonomic disturbance | ||
| Diphtheria—— | Cranial nerve onset | Demyelinative. No inflammatory infiltration. |
| Mixed motor/sensory | ||
| Porphyria—— | Motor (may begin in arm). | Axonal |
| Autonomic disturbance | ||
| Minimal sensory loss. | ||
| SUBACUTE – occasionally CHRONIC: months and years | ||
| ASYMETRICAL and MULTIFOCAL | ||
| Infections | ||
| Leprosy | Sensory neuropathy, often multifocal; associated depigmentation | Spectrum from paucibacillary (few organisms with intense inflammation) to multibacillary (many organisms with little inflammation) |
| HIV | Range of associated neuropathies | |
| Vasculitic disorders | ||
| Polyarteritis nodosa; | Usually presents with mononeuritis multiplex or an asymmetrical sensorimotor neuropathy. | Vasculitis with Wallerian degeneration in distal nerves |
| Wegner’s granulomatosis; | ||
| Churg-Strauss syndrome | ||
| Often painful | ||
| Non-systemic vasculitis | As above without systemic features | |
| SUBACUTE and CHRONIC: months and years | ||
| SYMMETRICAL | ||
| Metabolic and endocrine disorders | ||
| Diabetes | Most commonly distal sensorimotor | |
| But wide range of other forms (see later) | ||
| Ureamia | Distal sensorimotor | Axonal degeneration |
| Hypothyroidism | Distal sensorimotor | |
| Acromegaly | Distal sensorimotor | |
| CAUSE | FUNCTIONAL DISTURBANCE | PATHOLOGY |
|---|---|---|
| Nutritional deficiencies | ||
| Vitamin B1 (thiamine) | Predominantly sensory, with burning feet. | Axonal degeneration with segmental demyelination |
| Includes alcoholic neuropathy | Weakness may develop. | |
| Autonomic involvement common but mild | ||
| B12 deficiency | Predominantly sensory; may be associated spinal cord involvement | |
| Malignant disease | ||
| Paraneoplastic | Sensory or sensorimotor | Axonal; may be associated antibodies (anti-Hu) |
| Infiltrative | Multifocal, often a polyradiculopathy | More common with lymphoma |
| Paraprotein associated | ||
| Monoclonal gammopathy (IgG, IgA, IgM) | Sensorimotor neuropathy | Axonal with segmental demyelination |
| Chronic inflammatory demyelinating polyneuropathy (CIDP) (see later) | ||
| Amyloid | ||
| Primary, secondary or familial | Sensorimotor neuropathy often with autonomic involvement | Thickened nerves with amyloid deposition and small fibre neuropathy |
| May present as multiple mononeuropathies | ||
| Inherited neuropathies | ||
| Charcot-Marie-Tooth disease (see below) | ||
| Refsum’s disease | Phytanic acid storage disorder. Sensorimotor neuropathy with ichthyosis, retinitis pigmentosa and deafness | |
| Drug induced | ||
| Wide range of drugs induce neuropathies including: | ||
| Antibiotics | Metronidazole; ethambutol; isoniazid; nitrofurantoin; dapsone | |
| Oncology drugs | Adriamycin; cisplatin; taxanes; vincristine | |
| HIV drugs | Didanosine; stavudine; zalcitabine | |
| Others | Amiodarone; gold; phenytoin | |
| Toxin induced | ||
| Solvents | ||
| Heavy metals | Lead; arsenic; thallium | |
Chronic idiopathic axonal neuropathy:
INVESTIGATION OF NEUROPATHY
For a patient with a distal symmetrical sensorimotor neuropathy:
Further investigations (depending on clinical history):
THE POLYNEUROPATHIES – SPECIFIC TYPES
GUILLAIN BARRÉ SYNDROME (ACUTE INFLAMMATORY DEMYELINATING POLYNEUROPATHY)
Investigations
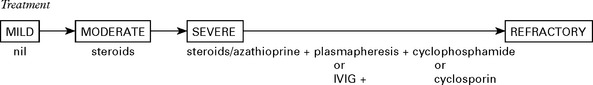
Treatment
Treatment is generally given to those who can no longer walk and is deferred in milder cases.
CHRONIC INFLAMMATORY DEMYELINATING POLYNEUROPATHY (CIDP)
Prevalence – 3% of all neuropathies
Age of onset: mean 35 yrs (fluctuating course – younger, progressive – older)
| Diagnosis: | Electrophysiology | |
| Distinguish from |
Outcome with treatment – 30% symptom free – 45% mild disability – 25% severe disability
DIABETIC NEUROPATHY
This condition is uncommon in childhood and increases with age.
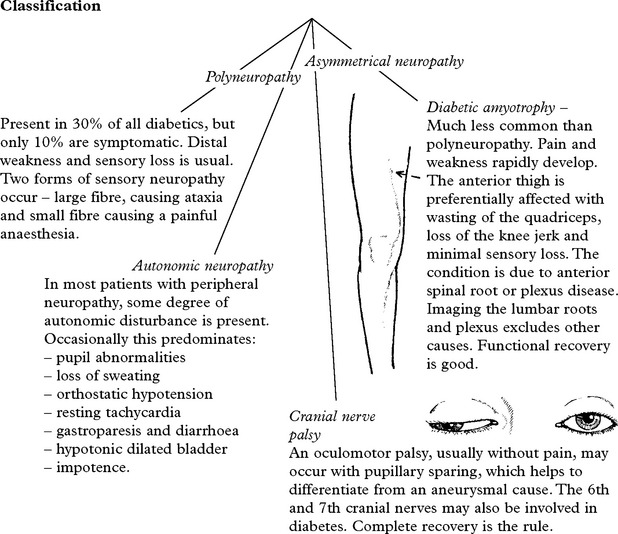
CARCINOMATOUS POLYNEUROPATHY
THE POLYNEUROPATHIES – SPECIFIC TYPES – INHERITED NEUROPATHIES

PLEXUS SYNDROMES AND MONONEUROPATHIES
Cranial nerve mononeuropathies have been dealt with separately.
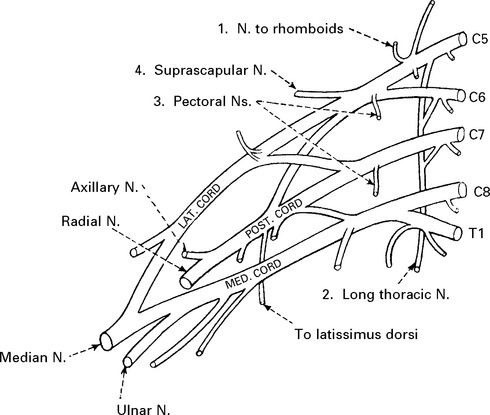
BRACHIAL PLEXUS SYNDROMES
UPPER PLEXUS LESION (C5C6)

When damage to C5C6 is more proximal, nerve to rhomboids and long thoracic nerve may be affected.

LOWER PLEXUS LESION (C8T1)
Forced abduction of the arm at birth (Klumpke’s paralysis) or trauma may produce damage to the lower plexus. This results in paralysis of the intrinsic hand muscles producing a claw hand, C8T1 sensory loss and a Horner’s syndrome (page 145) if the T1 root is involved.
TOTAL BRACHIAL LESION
This results in complete flaccid paralysis and anaesthesia of the arm.
The presence of a Horner’s syndrome indicates proximal T1 nerve root involvement.
THORACIC OUTLET SYNDROME
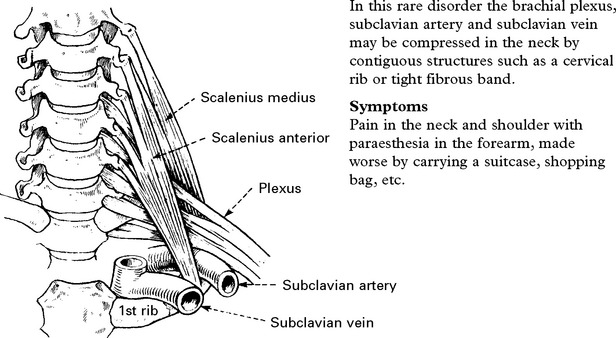
Signs
Sensory loss in a T1 distribution.
Wasting and weakness of thenar and occasionally interosseous muscles.
Signs of vascular compression:
BRACHIAL NEURITIS (Neuralgic amyotrophy)
Brachial neuritis is a relatively common disorder sometimes associated with:
In most cases it develops without any evident precipitating cause.
Clinical features
< div class='tao-gold-member'>
Related posts:
 CLINICAL PRESENTATION, ANATOMICAL CONCEPTS AND DIAGNOSTIC APPROACH
CLINICAL PRESENTATION, ANATOMICAL CONCEPTS AND DIAGNOSTIC APPROACH
 GENERAL APPROACH TO HISTORY AND EXAMINATION
GENERAL APPROACH TO HISTORY AND EXAMINATION
 LOCALISED NEUROLOGICAL DISEASE AND ITS MANAGEMENT B. SPINAL CORD AND ROOTS
LOCALISED NEUROLOGICAL DISEASE AND ITS MANAGEMENT B. SPINAL CORD AND ROOTS
 INVESTIGATIONS OF THE CENTRAL AND PERIPHERAL NERVOUS SYSTEMS
INVESTIGATIONS OF THE CENTRAL AND PERIPHERAL NERVOUS SYSTEMS
 LOCALISED NEUROLOGICAL DISEASE AND ITS MANAGEMENT A. INTRACRANIAL
LOCALISED NEUROLOGICAL DISEASE AND ITS MANAGEMENT A. INTRACRANIAL
 MULTIFOCAL NEUROLOGICAL DISEASE AND ITS MANAGEMENT
MULTIFOCAL NEUROLOGICAL DISEASE AND ITS MANAGEMENT
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


