Complex attention
The patient has increased difficulty in environments with multiple stimuli (e.g., TV, radio, conversation); has difficulty holding new information in mind (e.g., recalling phone numbers or addresses just given; or reporting what was just said).
Executive function
The patient is not able to perform complex projects; needs to rely on others to plan instrumental activities of daily living or make decisions.
Learning and memory
The patient repeats self in conversation, often within the same conversation; cannot keep track of short list of items when shopping or of plans for the day. Requires frequent reminders to complete task in hand.
Language
The patient has significant difficulties with expressive or receptive language; often uses general terms such as “the thing” and “you know what I mean.”
With severe impairment patients may not even recall names of close family and friends.
Perceptual–Motor
The patient has significant difficulties with previously familiar activities (e.g., using tools, driving motor vehicle), and navigating in familiar environments.
Social cognition
The patient may exhibit changes in behavior (e.g., shows insensitivity to social standards); makes decisions without regard to safety. Usually patients have little insight into these changes.
13.1.1 Epidemiology
By 2005, 24.2 million people worldwide had MNCD and 4.6 million new cases were arising every year (Ferri et al. 2005; Reitz and Mayeux 2014). It is estimated that the highest prevalence and incidence rates of MNCD can be found in North America and Western Europe, followed by populations in Latin America and China and the western-Pacific region (Ferri et al. 2005). Studies predict that global prevalence of MNCD will quadruple by the year 2050 (Reitz and Mayeux 2014). Much of the increase will be in the developing countries (Alzheimer’s Disease International 2013).
Studies have found that the prevalence of MNCD increases exponentially with age (Table 13.2) (Lobo et al. 2000; Alzheimer’s Disease International 2008). In 2002, the Aging, Demographics, and Memory Study (ADAMS) estimated the prevalence of MNCD in the USA among individuals aged 71 and older to be 14 %, comprising about 3.4 million individuals, and in those aged 90 and older 37.4 % (Plassman et al. 2007).
Table 13.2
Incidence and prevalence rates of dementia from the EURODEM meta-analyses for European studies
Age group | Annual incidence per 100 | Prevalence (%) | ||
|---|---|---|---|---|
Males | Females | Males | Females | |
60–64 | 0.2 | 0.2 | 0.4 | 0.4 |
65–69 | 0.2 | 0.3 | 1.6 | 1.0 |
70–74 | 0.6 | 0.5 | 2.9 | 3.1 |
75–79 | 1.4 | 1.8 | 5.6 | 6.0 |
80–84 | 2.8 | 3.4 | 11.0 | 12.6 |
85–89 | 3.9 | 5.4 | 12.8 | 20.2 |
90+ | 4.0 | 8.2 | 22.1 | 30.8 |
Overall, AD accounted for approximately 69.9 % of all dementia, while vascular dementia (VaD) accounted for 17.4 %. Other types of dementia such as “dementia, undetermined etiology,” Parkinson’s dementia, normal-pressure hydrocephalus, frontal lobe dementia, alcoholic dementia, traumatic brain injury, and Lewy body dementia accounted for the remaining 12.7 % of cases (Plassman et al. 2007). The Global Burden of Disease project DISMOD-II software estimated incidence rates of 4.6 million new cases of dementia every year (about one new case every 7 s). Estimating the number of people living with dementia worldwide in 2001 at 24.3 million, but predicting the number will almost double every 20 years, translates to 42.3 million in 2020 and 81.1 million in 2040 (Ferri et al. 2005).
MNCD has widespread effects on affected individuals, their families, and society. In the report on the state of US Health, Alzheimer’s Disease (AD) was ranked as the ninth cause of the years of life lost due to premature mortality and the 12th cause of the years lived with disability (Murray et al. 2013).
Early-onset neurocognitive disorder (NCD) has been increasingly recognized. A study conducted at the university hospital in Cambridge, UK found that the overall prevalence of early-MNCD (i.e., onset <65 y/o) was 81 per 100,000 in the 45–64-year age group (Ratnavalli et al. 2002). Furthermore, they found that AD accounted for 35 % of this early onset dementia, while frontotemporal lobar degeneration (FTLD) accounted for 22 %. As mentioned above, there are multiple etiologies contributing to the dementias, each with its own characteristics and presentation. The commonest subtypes of MNCD include AD, vascular dementia (VaD), dementia with Lewy bodies, and FTLD. DSM-5 subtypes the various MNCD syndromes based on their etiology, if known (Table 13.3) (APA 2013). These are discussed below.
Table 13.3
Major neurocognitive disorders—subtypes (as recognized by DSM-5)
Subtypes based on etiology (in alphabetical order) |
Alzheimer’s disease |
Due to another medical condition |
Due to multiple etiologies |
Frontotemporal lobar degeneration |
HIV infection |
Huntington’s disease |
Lewy body disease |
Parkinson’s disease |
Prion disease |
Substance/medication induced |
Traumatic brain injury |
Unspecified |
Vascular disease |
Subtypes based on severity level |
Mild—Instrumental ADL’s are preserved |
Moderate—Basic ADL’s affected |
Severe—Fully dependent |
Subtypes based behavior |
With behavioral disturbance |
Without behavioral disturbance |
13.2 Etiologic Factors and Subtypes
13.2.1 Alzheimer’s Disease
Alzheimer’s disease (AD) is the leading cause of MNCD, estimated to occur in 50–75 % of those afflicted by dementia (Gouras 2009). It is estimated that by 2013 about five million Americans older than 65 have Alzheimer’s disease. Further estimates suggest that by the year 2025 up to 7.1 million of Americans older than 65 will suffer from the condition. The disease slowly erodes memory and thinking skills, and eventually makes the affected individual incapable of taking care of their activities of daily living (ADLs). It is characterized by slow progression with an average time from its diagnosis until death ranging from 5 years (Larson et al. 2004) to 10 years (Brookmeyer et al. 2002). MNCD due to AD usually presents with loss of recent episodic memory, with affected individuals becoming forgetful, losing objects, repeating stories, and missing appointments. Word-finding difficulty is common and presents early. Memory deficits are followed months to years by deficits in executive function, visuospatial function, language, and praxis. It eventually manifests in global cognitive deterioration, difficulty with long-term memory and overlearned visuospatial tasks, such as eating and dressing (Table 13.4).
Table 13.4
Major neurocognitive disorder due to Alzheimer’s disease
1. Diagnostic criteria for major neurocognitive disorder are met. |
2. There is insidious onset and gradual progression of impairment in one or more cognitive domains. |
3. Criteria met for either probable or possible Alzheimer’s disease are as follows: |
(a) Probable Alzheimer’s disease is diagnosed if either of the following is present: |
• Evidence of a causative Alzheimer’s disease genetic mutation from family history or genetic testing. |
• All three of the following are present: |
– Clear evidence of decline in memory and learning and at least one other cognitive domain (based on detail history or serial neuropsychological testing). |
– Steadily progressive, gradual decline in cognition, without extended plateaus. |
– No evidence of mixed etiology (e.g., absence of other neurodegenerative or cerebrovascular disease or another neurological, mental or systemic disease likely contributing to cognitive decline). |
(b) Otherwise, possible Alzheimer’s disease should be diagnosed. |
Of note, behavioral changes are common and eventually up to 88 % of patients experience NCD-associated behavioral and psychiatric symptoms, formerly known as Behavioral and Psychiatric Symptoms of Dementia (BPSD) (Mega et al. 1996). These symptoms are usually differentiated into three clusters, i.e., affective symptoms (dysphoria, anxiety, apathy), psychotic symptoms (delusions and hallucinations), and verbal and physical agitation. Of note, psychotic symptoms are different from the psychotic symptoms in primary psychotic illnesses: hallucinations are more commonly visual and delusions often relate to the faulty memories. For example, a patient misplaces items and thus concludes that there are intruders in the house who steal, or they have difficult time remembering their spouse and conclude that he/she was replaced with an impostor (Capgras delusion). Usually these behavioral symptoms bring these patients to the attention of a CL psychiatrist and management of agitation in a patient with MNCD is not an uncommon consult question in the general hospital. These neuropsychiatric symptoms also place a huge burden on the family and are frequently the reason behind nursing home placement of such patients. Eventually patients are bedridden, unable to feed themselves or mobilize and they often die from dehydration or sepsis.
The greatest risk factor for developing AD is aging; in addition, history of head trauma and small head size (McDowell 2001) also increase this risk. Higher level of education and occupational attainment may be protective factors (Ngandu et al. 2007), although studies supporting this might have had multiple confounders.
13.2.1.1 Pathology
The “amyloid hypothesis” is at the core of the demonstrated neuropathology of AD development. It proposes that an overproduction and decreased degradation of β-amyloid (Aβ) protein lead to cerebral amyloid angiopathy (CAA), which is characterized by progressive loss of smooth muscle cells in arterioles and accumulation of eosinophilic hyaline material, and formation of senile plaques (SPs), which are comprised of the core of amorphous eosinophilic globule of amyloid surrounded by neuritic corona (Fig. 13.1). Several lines of evidence support that Aβ accumulation precedes and can induce tangle pathology (Gouras 2009). Amyloid precursor protein (APP) is a precursor to β-amyloid and is coded on chromosome 21. Further support comes from the fact that nearly all persons with trisomy 21 (Down’s Syndrome) who live long enough develop AD pathology and an accompanying behavioral syndrome. Moreover, mutations in the APP gene lead to the early-onset AD (Goate et al. 1991) as do mutations in two additional identified genes (PSEN1 and PSEN2) (Lippa et al. 2000).
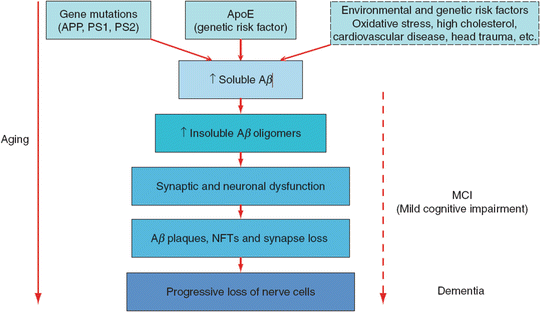
Fig. 13.1
The amyloid cascade hypothesis. APP amyloid precursor protein, PS1, PS2 presenilin proteins 1 and 2, Apo E apolipoprotein E, Ab b-amyloid, NFTs neurofibrillary tangles. From (Gouras 2009 p. 405)
In addition, there has been an established association between polymorphisms of the apolipoprotein E gene (APOE), pleiotropic protein with effects on neurotoxicity, tau phosphorylation, synaptic plasticity, and inflammation, and the risk of onset of AD at least in white populations (Corder et al. 1993). The greatest risk of AD with earlier age of onset is associated with presence of two copies of ε4 allele; presence of ε3 carries a diminished risk, and ε2—even lower risk.
In addition to senile plaques, the microscopic lesions of AD include neurofibrillary tangles (NFTs), which are dense intraneuronal cytoplasmic aggregates of paired helical filaments and granulovacuolar degeneration (GVD), characterized by neuronal cytoplasm of hippocampal pyramidal cells replaced by vacuoles with small basophilic granules. Moreover, there is significant synapse loss that can be demonstrated biochemically or by immunohistochemistry in AD brain.
13.2.1.2 Diagnosis
Patients presenting with impairing cognitive or behavioral symptoms should be evaluated carefully and diagnosis of MNCD, and in particular AD, should be considered. Repetition is important to rule out other reversible causes of cognitive impairment, such as etiologies contributing to delirium (infection, medication side effect), hypothyroidism or hyperthyroidism, cobalamin deficiency, and neurosyphilis in some geographical regions. Computed Tomography (CT) brain scan can help to identify significant brain pathology for which interventions might be possible (brain tumor, hydrocephalus, subdural hematoma, large stroke). Brain Magnetic Resonance Imaging (MRI) can provide additional information about subtle white matter ischemic changes and regional cerebral atrophy pattern. It is important to screen for depression and other psychiatric morbidity, as these etiologies can contribute or solely explain the presentation. However, these symptoms might also be a part of MNCD. Cognitive testing must be done, such as the Mini Mental State Examination (MMSE) or the Montreal Cognitive Assessment (MOCA) (see Chap. 4 Appendix for these tests) and if not possible or not enough, formal neuropsychological testing is recommended.
The imaging of the AD patient brain with head CT scan or brain MRI demonstrates cerebral atrophy globally, but in particular in medial temporal lobes. Nuclear Imaging with fluorodeoxyglucose positron emission tomography (FDG-PET) (Benson et al. 1983) or single-photon emission computed tomography (SPECT) (Jagust et al. 1987) shows hypometabolism or hypoperfusion in temporoparietal regions with the sensitivity of 94 % and specificity of 73 % in pathologically proven AD (Silverman et al. 2001). Because this pattern of hypometabolism is so distinctive from that of FTD, distinguishing patterns of hypometabolism between AD and FTD is Medicare-approved indication for nuclear imaging.
13.2.2 Frontotemporal Lobar Degeneration
Frontotemporal Lobar Degeneration (FTLD, frontotemporal dementia, FTD) is a heterogeneous group of conditions with prominent early behavioral disinhibition, encompassing a variety of clinical syndromes and pathological substrates. The mean age at onset of FTLD is 52.8 years and there is a striking male preponderance (14:3). (Ratnavalli et al. 2002)
FTLD is usually divided into three clinical variants. These include the frontal-variant or behavioral-variant (fvFTLD); progressive nonfluent aphasia (PNFA); and semantic dementia (SD). The motor syndromes of corticobasal degeneration (CBD); progressive supranuclear palsy (PSP); and motor neuron disease (MND) may also be associated with features of FTLD and its pathology (Weder et al. 2007).
FTLD presents earlier as compared to AD with age of onset varying from 35 to 75, but most typically in 5th and 6th decades. It represents 20 % of degenerative dementias of pre-senile onset. Up to 40 % of patients have family history of FTLD, with another prominent risk factor being history of head trauma (Weder et al. 2007). Median survival from symptom onset is approximately 6 years for FTLD and 3 years for FTLD-MND (Hodges et al. 2003; Weder et al. 2007). Median survival for the entire group is 3 years from initial diagnosis, related to common significant delay in diagnosis.
Patients present with behavioral alterations and tend to lack appropriate basic and social emotions. Some patients with FTLD present with disinhibition and overactivity, while others show apathy and blunted affect.
Frontal variant of FTLD (fvFTLD) is characterized by insidious onset of personality changes, behavioral abnormalities and poor insight (Weder et al. 2007). Symptoms include disinhibition, poor impulse control, antisocial behavior, and stereotyped/perseverative behaviors. Patients might be interpersonally inappropriate, tactless, and offer and display improper sexual comments and gestures. Apathy and emotional blunting are common. Speech output is attenuated and mutism eventually develops. The most common cognitive deficit in fvFTD is an impairment of executive function or working memory, with other frequently encountered cognitive abnormalities including attentional deficits, poor abstraction, difficulty shifting mental set, and perseverative tendencies. Deficits in planning, organization and other aspects of executive function become universal as the disease progresses.
Semantic dementia (SD) or temporal FTD is associated with bilateral atrophy of the middle and inferior neocortex and is characterized by a loss of word meaning/knowledge. Patients present with abnormal speech, where speech is fluent, but words might be substituted for less specific ones and patients are often unaware of their difficulties with comprehension. Patients lose the ability to name and understand words and to recognize the significance of faces, objects and other sensory stimuli. In addition, they might demonstrate deficits on nonverbal tasks using visual, auditory, and other modalities. Behavioral symptoms may appear early or late.
Progressive nonfluent aphasia (PNFA) is associated with asymmetric atrophy of left hemisphere and is characterized by agrammatic nonfluent speech and decreased speech output leading to mutism. Patients present with changes in fluency, pronunciation, or word finding difficulty. Behavioral problems appear later in the disease.
13.2.2.1 Pathology
Pathology of FTD is heterogeneous and is characterized by gliosis, neuronal loss, and superficial spongiform degeneration in the frontal and/or temporal cortexes. Ballooned neurons, i.e., Pick cells, occur with variable frequency in all subtypes (Kertesz and Munoz 2002). Some cases show tau- or ubiquitin-positive inclusions, or lack any distinctive histological features (Mariani et al. 2006). Mutations in the tau gene, which is involved in the regulation of microtubule assembly and disassembly, lead to tau deposition in neurons and glia; while mutations in the progranulin gene lead to ubiquitin-only immunoreactive inclusions. Both genes are located on chromosome 17.
Neuroimaging shows anterior temporal and frontal atrophy, while functional imaging shows decreased perfusion of both frontal and temporal lobes. The focus of the atrophy is in the left temporal lobe in progressive nonfluent aphasia (PNFA) patients and in both frontal lobes in frontal variety frontotemporal dementia (FvFTD) patients.
Neurochemical changes of FTD differ from those of AD. There is evidence of less cholinergic deficit and more serotonergic disturbance in FTD as compared to AD (Weder et al. 2007). This might explain early increased impulsivity, irritability, affective change, and changes in eating behavior in patients with FTD since these behaviors are modulated by serotonergic dysfunction. Thus, serotonergic agents might have a greater role in managing behavioral symptoms of FTD as compared to cholinergic medications, as is discussed later in the chapter.
13.2.3 Vascular Disease
Vascular disease (VaD) encompasses a variety of vascular etiologies, including multi-infarct MNCD with cortical and subcortical involvement as well as the smaller lacunar and micro-infarcts (Erkinjuntti 2007).
VaD is considered the second most common cause of MNCD accounting for 10–50 % of the cases, with prevalence ranging from 1.2 to 4.2 % in persons aged 65 years and older (Hebert and Brayne 1995). The pathophysiology is attributed to interactions between vascular etiologies (coronary vascular disease and vascular risk factors, such as hypertension, diabetes, smoking), changes in the brain (infarcts, white matter lesions (WMLs), atrophy), and host factors (age, education) (Erkinjuntti 2007). The main subtypes of VaD include cortical VaD or multi-infarct MNCD also referred as post-stroke VaD, subcortical ischemic vascular disease (SIVD) or small-vessel MNCD, strategic-infarct MNCD, and hypoperfusion MNCD resulting from global cerebrovascular insufficiency (Erkinjuntti 2007).
Cortical VaD (multi-infarct MNCD, post-stroke VaD) is characterized by a relatively abrupt onset (days to weeks), a stepwise deterioration (some recovery after worsening) and a fluctuating course of cognitive functions. It is related predominantly to large vessel disease and cardiac embolic events. The frequency of post-stroke MNCD varies from 12 to 32 % within 3 months to 1 year after stroke (Leys et al. 2005). A history of stroke increases the risk of subsequent MNCD by a factor of 5 (Leys et al. 2005).
The presenting symptoms include memory impairment, which may be mild, and such cortical symptoms as aphasia, apraxia, agnosia, and visuospatial or constructional difficulty. In addition, most patients have some degree of dysexecutive syndrome. Moreover, patients often have focal neurological impairments apparent on the exam such as visual field deficits, lower facial weakness, focal motor or sensory deficits, and gait impairment (Leys et al. 2005).
Subcortical ischemic vascular Dementia (SIVD) or small-vessel MNCD incorporates two entities, “the lacunar state” and “Binswanger’s disease.” The onset is variable with 60 % of the patients having a slow onset and only 30 % an acute onset of cognitive symptoms (Erkinjuntti 2007). The course is gradual without (40 %) and with (40 %) acute deficits, and fluctuating in only 20 % (Babikian and Ropper 1987). There is often a preceding clinical history of transient ischemic attacks with only mild focal findings (e.g., drift, reflex asymmetry, gait disturbance). SIVD is attributed to small-vessel disease and is characterized by lacunar infarcts, focal and diffuse ischemic white matter lesions (WMLs), and incomplete ischemic injury. Clinically, it is characterized by the subcortical cognitive syndrome with deficits in executive functioning, slowed information processing, mild memory deficits and behavioral symptoms such as depression and emotional lability (Babikian and Ropper 1987). In addition, neurologic symptoms include motor hemiparesis, bulbar signs and dysarthria, and gait disorder. Imaging reveals multiple lacunes and extensive WMLs.
Strategic-infarct MNCD is characterized by focal, often small, ischemic lesions involving specific sites critical for higher cortical functions, such as the hippocampal formation, angular gyrus and cingulate gyrus, and subcortical sites leading to impairment, including thalamus, fornix, basal forebrain, caudate, globus pallidus, and the genu or anterior limb of the internal capsule (Erkinjuntti 2007).
Moreover, AD and vascular disease coexist in a large proportion of patients, making at times distinguishing primary etiology of MNCD difficult (Erkinjuntti 2007).
13.2.4 Lewy Body Disease
Lewy Body Disease (LBD, DLB) represents up to 20 % of all cases of MNCD cases. It presents late, in 6th through 9th decade and affects both genders equally (Ferman and Boeve 2007). The MNCD is characterized by cortical and subcortical cognitive impairments, with worse visuospatial and executive dysfunction as compared to AD. There is usually relative sparing of memory especially early on. Fifty percent of the cases have mixed presentation with AD. The diagnosis is based on presence of MNCD and additional two out of three features: spontaneous parkinsonism, hallucinations, and daily fluctuation in cognition. Parkinsonian signs must be spontaneous and not attributable to neuroleptics; as compared to Parkinson’s Disease (PD) or Parkinson’s Disease Dementia (PDD), there is more rigidity and bradykinesia than tremor and parkinsonian symptoms are less severe. Tremor, bradykinesia, and rigidity tend to be more symmetric than asymmetric, and tremor tends to be maximal with posture/action rather than at rest (Ferman and Boeve 2007).
Visual hallucinations (VH) in Dementia with Lewy Bodies (DLB) consist of fully formed, detailed, three-dimensional objects, people, or animals. Auditory hallucinations might happen, but mostly in patients who also have VH. VH have been documented to occur in 59–85 % of autopsy-confirmed DLB samples as compared to 11–28 % of autopsy-confirmed AD sample (Ferman and Boeve 2007). The etiology of DLB hallucinations is likely multifactorial, including severe depletion of acetylcholine, depletion of other neurotransmitters such as dopamine and serotonin, as well as intrusion of dream imagery into wakefulness as a potential mechanism due to the dysregulation of rapid eye movement (REM) sleep in many patients with DLB.
The fluctuations of DLB are characterized by a waxing and waning of cognition, abilities, and arousal. Moreover, patients who have DLB often have daytime drowsiness or somnolence. In addition, these patients often have the parasomnia of REM sleep behavior disorder (RBD) due to the loss of normal muscle atonia during REM. The augmented muscle activity during REM sleep occurs along with dream content and can range from elevated muscle tone to complex behavioral sequences.
Of note, REM sleep behavior disorder can precede the onset of neurodegenerative diseases with alpha-synuclein inclusions (i.e., DLB, PD, or multiple system atrophy [MSA]) by years and even decades and is postulated to be a precursor to the disorders (Iranzo et al. 2013).
In addition, autonomic abnormalities, in particular orthostatic hypotension and carotid sinus sensitivity, are more common in DLB than AD or elderly controls.
To make a diagnosis of DLB, please refer to criteria to make a clinical diagnosis (Table 13.1).
13.2.4.1 Pathology
Neuropathologically, DLB is marked by presence of Lewy bodies in cortical and neocortical brain regions. Lewy bodies, typically present in Parkinson’s Disease (PD), are concentric, intracytoplasmic neuronal inclusions within monoaminergic and cholinergic neurons of the substantia nigra, locus ceruleus, and basal nucleus of Meynert with dense eosinophilic core surrounded by a lucent halo, easily seen with routine staining. In contrast to PD, the neocortical Lewy bodies seen in DLB are smaller, lack a halo, and are difficult to see under routine staining conditions. PD and cortical Lewy bodies contain a-synuclein, which is a 140 amino acid protein of unknown function. Abnormal protein processing gives rise to the cytoplasmic collections of a-synuclein, which coalesce to form Lewy bodies. Brains of patients with Lewy bodies demonstrate severe depletion of both cholinergic and dopaminergic markers (Walker et al. 2007).
13.2.5 Posttraumatic Brain Injury
Chronic traumatic encephalopathy (CTE) is a progressive neurodegenerative disease that occurs in association with repetitive traumatic brain injury (McKee et al. 2014). In most instances, the clinical symptoms of the disease begin after a long period of latency ranging from several years to several decades. The initial symptoms are typically insidious, consisting of irritability, impulsivity, aggression, depression, short-term memory loss, and heightened suicidality. The symptoms progress slowly over decades to include cognitive deficits and MNCD. MNCD has been increasingly associated with traumatic brain injuries. Mildcognitive impairment (MCI) after self-reported head trauma with at least momentary loss of consciousness or memory has been found to be associated with greater amyloid deposition, when compared with cognitively intact individuals, suggesting that head trauma may be associated with the development of MNCD (Mielke et al. 2014).
The underlying pathology of CTE is characterized by the accumulation of phosphorylated tau protein in neurons and astrocytes in a pattern that is unique from other tauopathies, including Alzheimer’s disease (McKee et al. 2014). The hyper-phosphorylated tau abnormalities begin focally, as perivascular neurofibrillary tangles and neurites at the depths of the cerebral sulci, and then spread to involve superficial layers of adjacent cortex before becoming a widespread degeneration affecting medial temporal lobe structures, diencephalon and brainstem. Most instances of CTE (>85 % of cases) show abnormal accumulations of phosphorylated 43 kDa TAR DNA binding protein that are partially colocalized with phosphorylated tau protein. In addition CTE is associated with frontal and temporal lobe atrophy, and is increasingly categorized as an acquired Frontotemporal lobar degeneration. Clinically CTE is characterized by behavioral and personality changes, as well as cognitive impairments. As is the case in AD, at present CTE cannot be definitively diagnosed during life, thus its exact incidence and prevalence remain uncertain.
13.2.6 Rapidly Progressive Dementias
Rapidly Progressive Dementias (RPDs) are neurologic conditions that develop subacutely over weeks to months, sometimes even days, not infrequently quickly leading to death (Geschwind et al. 2007). The differential is extensive and is demonstrated along with suggested workup in Table 13.5.
Table 13.5
Differential and suggested workup for rapidly progressive dementias
Category | Differential | Suggested tests |
|---|---|---|
Neurodegenerative | • AD, DLB, FTD • Neurofilament Inclusion Body Disease (NIBD) • Fahr’s Disease | • Brain MRI • Fluorodeoxyglucose Positron Emission Tomography (FDG-PET) scan |
Infectious | • Viral encephalitis, e.g.. HSV • HIV dementia • Progressive Multifocal Leukoencephalopathy (PML) (JC virus) • Opportunistic infections in immunocompromised: Cryptococcus, mycobacteria • Neurosyphilis • Subacute Sclerosing Panencephalitis (SSPE) (measles virus) | • Viral PRCs and cultures • Bacterial, fungal, AFB stains and cultures • RPR • Whipple’s PCR |
Autoimmune | • Non-vasculitis autoimmune inflammatory meningoencephalopathies: primary angiitis of the CNS (PACNS), polyarteritis nodosa (PAN), sarcoidosis, Systemic Lupus Erythematosus (SLE), Sjögren’s syndrome, celiac disease, Behçet’s disease, hypereosinophilic syndrome • Cerebral amyloid inflammatory vasculopathy • Hashimoto’s encephalopathy • Limbic encephalitis | • ESR, CRP, C3, C4, ANA, rheumatoid factor, anti-SSA, anti-SSB, anti-dsDNA, anti-smith, P-ANCA, C-ANCA, anti-endomysial or anti-gliadin IgA or IgC, SSA, SSB, ACE • TSH, free T4, anti-thyroid peroxidase • Paraneoplastic panel (CSF and serum) |
Malignant | • Primary and metastatic solid tumors • Primary CNS lymphoma (PCNSL) • Intravascular lymphoma (i.e., angiotropic lymphoma) • Lymphomatoid granulomatosis | • CT scan body with and without contrast • Whole body PET scan • CSF cytology and flow cytometry • Serum LDH, tumor markers (PSA, CEA, etc.) • Mammogram • Colonoscopy |
Vascular | • Strokes • Thrombotic thrombocytopenic purpura (TTP) • Hyperviscosity syndromes: polycythemia vera, gammopathies • CNS vasculitidies | • Brain imaging • Hypercoagulability testing; coagulation profile • Echocardiogram; carotid ultrasound • Cerebral angiogram, meningeal biopsy |
Toxic-metabolic | • Vitamin B1, B12, niacin, folate deficiencies • Uremia • Wilson’s disease • Portosystemic encephalopathy • Acquired hepatocerebral degeneration • Porphyria • Bismuth, lithium, mercury, arsenic toxicities • Electrolyte abnormalities | • Vitamin B1, B12, niacin, folate • Comprehensive metabolic panel: electrolytes, liver function tests, creatinine/ blood urea nitrogen • Copper and ceruloplasmin; 24 h copper • 24 h urine heavy metal for lead, arsenic, mercury, bismuth, albumin, lithium • Methylmalonic acid levels; thiamine, vitamin E • Exposure history |
Prion | • Creutzfeldt–Jakob Disease (CJD) | • EEG • CSF cytology, including protein 14-3-3 • Brain MRI • Brain biopsy |
Table 13.6
Mechanisms mediating delirium and cognitive impairment
1. A number of factors and mechanisms leading to delirium, may also directly cause CNS damage and neuronal dysfunction, and thus mediate both the manifestations of delirium and long-term cognitive impairment (e.g., cytokine release and other neuroinflammatory mediators; decrease perfusion and oxygenation leading to decreased cerebral oxidative metabolism; changes in blood–brain barrier permeability; hypercatabolic states; water and electrolyte imbalances; excessive glucocorticoid levels and other HPA axis dysfunctions; melatonin and sleep–wake cycle abnormalities). |
2. Pharmacological agents used either to treat the underlying causes of the delirium (e.g., steroids, calcineurin inhibitors, other immunosuppressants, dopamine) or those agents used to treat delirium (e.g., dopamine blocking agents, benzodiazepines) may themselves lead to neuronal damage in a fragile brain. |
3. Any of the mechanisms listed above may themselves lead to alterations in neurotransmitter concentration or receptor sensitivity which may underlie the different symptoms and clinical presentations of delirium and/or long-term cognitive dysfunction. Thus, the same mechanisms that cause the substrate for delirium, may mediate the cognitive impairments observed after the acute presentation of delirium has resolved. |
4. It is possible that instead of causing cognitive deficits or dementia, delirium (and its underlying causes) only serve as a catabolic agent, leading to an acceleration of normal physiological cerebral aging mechanisms leading to dementia. |
5. It is also possible that an episode of delirium simply unmasks subtle cognitive deficits already present, although not yet identified. |
One important category in this class is prion diseases, including Creutzfeldt–Jakob Disease (CJD), characterized by a classic triad of dementia, typical EEG changes, and myoclonus. Most cases are sporadic, with genetic cases comprising 15 % and iatrogenic 2 % (Eggenberger 2007). Prevalence, annual incidence, and yearly mortality of CJD are 0.5–1 per million people. It results from abnormal prion protein form acting in an “auto”-enzymatic fashion, converting normal host prion (prior protein cellular (PrPC)) into the abnormal isoform (protease resistant scrapie form of PrP (PrPSc)) (Eggenberger 2007). The onset of the illness is typically between 50 and 70 years of age with median age of onset at 68 and equal gender distribution. Median survival is 5 months and 85 % of afflicted die within first year of symptom onset. The onset is usually insidious with a nonspecific prodrome in one third, characterized by headache, fatigue, anxiety, changes in sleep, anorexia, weight loss, dizziness, memory difficulties, mood or behavior changes, weakness, and problems with locomotion. These symptoms are followed by progressive aphasia, apraxia, pyramidal signs, myoclonus, and choreiform-athetoid movements. Patients become severely demented within 6 months, with death occurring usually within 12 months of the symptom onset, typically resulting from intercurrent infection. Heidenhain variant is punctuated by visual presentation, most commonly a homonymous visual field defect leading to cerebral blindness early in the course of the disease. EEG can be helpful with the diagnosis, first showing slowing, and later in the course characterized by periodic sharp waves in two thirds of the patients. CSF studies might be remarkable for increased protein 14-3-3, total tau (t-tau), and neuron specific enolase (NSE). Brain MRI demonstrates increased bilateral signal intensity in the basal ganglia, corpus striatum, or thalamus, better visualized on diffusion-weight imaging (DWI) than on fluid-attenuated inversion recovery (FLAIR) sequences (Eggenberger 2007). The definitive diagnosis can be obtained via brain biopsy with tissue pathology demonstrating spongiform degeneration, astrocytic gliosis with neuronal loss, amyloid plaques, lack of inflammatory response, and misfolded prion proteins (PrPs) on immunochemistry (Yung et al. 2010).
Related posts:
 The Why and How of Psychiatric Consultation
Interviewing in Consultation-Liaison Psychiatry
Systems and Ethical Issues in CL Psychiatry: Hospital as a Social System, Sick Role and Doctor Role, Ethical and Legal Issues
Somatic Symptom and Related Disorders
Chronic Conditions, Lung Disease, Cancer, the Palliative Care Settings, and the Dying Patient
The Renal Dialysis and Kidney Transplant Patient
The Why and How of Psychiatric Consultation
Interviewing in Consultation-Liaison Psychiatry
Systems and Ethical Issues in CL Psychiatry: Hospital as a Social System, Sick Role and Doctor Role, Ethical and Legal Issues
Somatic Symptom and Related Disorders
Chronic Conditions, Lung Disease, Cancer, the Palliative Care Settings, and the Dying Patient
The Renal Dialysis and Kidney Transplant Patient
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


