SDF-1 [9]
IL-10 [10]
Nitrite/NO bioactivity [12]
microRNA 144 [11]
Cerebral Blood Flow (CBF) after RIC
In the cardiac studies of RIC, coronary blood flow is seldom directly measured. Many cardiac studies of RIC use the Langendorff heart perfusion model, an in vitro technique where the heart is removed and reverse perfused through the aorta, negating any ability to study regulation of coronary blood flow. However, in other model systems, there is evidence that RIC improves coronary blood flow. In a pig model of LAD occlusion, coronary resistance was decreased and coronary blood flow was increased by remote ischemic preconditioning [13]. Moreover, in healthy subjects and humans with congestive heart failure (CHF), RIC increased coronary microvascular perfusion as measured by transthoracic Doppler echocardiography [14].
In some of the earliest studies of preconditioning in the brain, improvements in microvascular perfusion and CBF were shown to contribute to the protective effect. Administration of lipopolysaccharide (LPS) protects against permanent MCA occlusion 72 h later with a reduction in infarct size. Microvascular flow was increased at 4 and 24 h after MCA occlusion in the LPS-treated mice compared with control mice, indicating that preservation of blood flow partially accounted for the protective effect [15].
In a permanent occlusion model, Zhao and Nowak [16] showed that ischemic preconditioning 24 h prior to permanent ischemia increased CBF and reduced infarct size in accord with preservation of local CBF. Brief episodes of ischemia (15 min) protected against a subsequent 45-min duration of ischemia in mice and increased CBF as measured by laser Doppler and MRI arterial spin labeling (ASL), demonstrating the critical importance of CBF as a mediator of protection [17].
In an autologous thromboembolic clot model in the mouse, we found that RIC increased CBF as measured by laser Doppler flowmetry and laser contrast speckle imaging (LCSI) in mice treated with tissue plasminogen activator (tPA) and in mice not treated with IV tPA [18–20]. This effect was seen in young males, ovariectomized females, and aged male mice (12 months old). We have also found that remote ischemic postconditioning daily for 2 weeks increases CBF in a mouse bilateral carotid artery stenosis (BCAS) model (Fig. 1) [21]. This increase in CBF persists at least 1 week beyond the cessation of remote limb postconditioning.
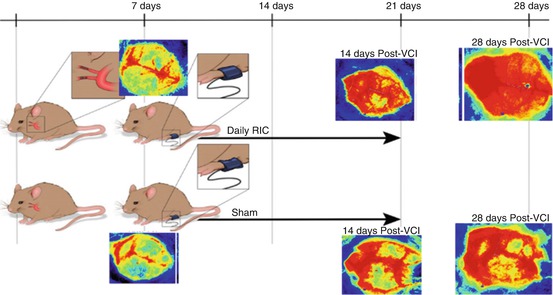
Fig. 1
Mice had microcoils applied to both internal carotid arteries. After 1 week, CBF was measured by laser contrast speckle imaging (LCSI) and showed a severe drop in CBF. Mice were randomized to RIC (top) or sham RIC daily (bottom) for 2 weeks. CBF was measured 21 and 28 days after placement of the coils. CBF was improved in the RIC mice at 21 and 28 days (7 days after stopping of RIC) (see Khan et al. [21] for details)
Nitric Oxide
Early work in preconditioning implicated NO in the mechanism of action. In a study of hypoxic preconditioning in a neonatal rodent hypoxia ischemia (HI) model, previous hypoxia (8 % oxygen) for 3 h completely protected against a subsequent HI-induced injury 24 h later. This was dependent on NO as the effect was abolished with L nitroarginine, a nonselective inhibitor of NOS but not with the selective nNOS (7-nitroindazole) or iNOS inhibitor (aminoguanidine), suggesting that preconditioning was dependent on eNOS (NOS3) [22]. In a permanent middle cerebral artery (MCA) occlusion model, brief occlusion of the MCA for 5 min repeated 3 times reduced infarct size in wild-type mice but not in eNOS or nNOS knockout mice indicating a role for NO and implicating both eNOS and nNOS [23]. NO generation via eNOS or nNOS is also required for the preconditioning effect of LPS [24].
In RIC, the NO system also appears to play a role in the mechanism of protection. In a mouse liver ischemia-reperfusion model, NO plays an essential role in mediating protection from RIC. Mice treated with RIC are protected from liver ischemic damage and have elevated microvascular blood flow. This protection is abolished with PTIO, a NO scavenger [25]. The mice treated with RIC have elevated NOx levels in the blood. Moreover, eNOS knockouts are not protected by RIC in this model [26].
NO and nitrite are also a key signaling pathway mediating RIC-induced cardioprotection. Rassaf et al. [12] reported that RIC protected hearts during 30-min LAD occlusion and increased nitrite levels in the plasma and hearts in mice. Nitrite levels also increased in the plasma of healthy human volunteers during RIC of the arm. The nitrite increase was dependent on reactive hyperremia during the reperfusion phase and related to shear stress-induced upregulation of eNOS. Cardioprotection with RIC in mice was lost in eNOS knockouts. Transfer experiments in the Langendorff heart model showed that plasma from conditioned humans was protective. Administration of exogenous nitrite was protective in mice. Myoglobin was essential to the protection, as myoglobin knockouts were not protected and myoglobin appeared to mediate nitrite reductase activity in the heart. These data meet the criteria to establish plasma nitrite as a mediator of RIC according the Working Group of the Cellular Biology of the Heart of the European Society of Cardiology [27].
In the mouse BCAS model where there are increases in CBF with chronic daily RIC, we measured plasma nitrite by ozone-chemiluminesence (GE Sievers NOA 280) in mice subjected to sham coiling, to mice with coiling and sham RIC, and in mice with BCAS with RIC (N = 6 per group) at 28 days post coiling. Mice subjected to BCAS had reduced plasma nitrite but RIC administered daily for 2 weeks dramatically and significantly increased plasma nitrite (Fig. 2). This increase was associated with increased CBF by LSCI in these mice and improved cognitive outcome.
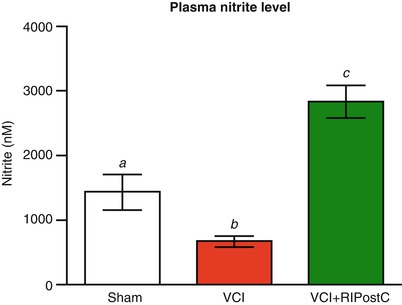
Fig. 2
Mice were randomized to BCAS surgery (as in Fig. 1) or to sham surgery. The BCAS mice (model for vascular cognitive impairment (VCI)) were randomized to RIC or sham RIC (N-6 per group). Plasma nitrite (measured at 28 days) was reduced in the mice with BCAS (b) compared with those with sham surgery (a), and there was a significant increase in mice randomized to RIC (c) compared with sham RIC (b) (p < .05)
Nitrite serves a “storage” pool of NO derived from endogenous eNOS that circulates in the blood associated with RBC/ hemoglobin and is reduced to NO in areas of hypoxemia, mediating hypoxic vasodilatation [28, 29]. As nitrite is a known vasodilator and increases CBF, upregulation of nitrite by RIC may explain the increase of CBF. Nitrite also is involved in the nitrosylation of key mitochondria proteins including complex I [12]. Therefore, increases of plasma nitrite likely increase CBF and protect mitochondria from oxidative stress.
Conclusions
Biomarkers of the conditioning response are needed in clinical trials of RIC to help select dose and duration of therapy. We propose that CBF as measured by MRI ASL and plasma nitrite could serve as imaging and blood biomarkers in clinical trials of RIC in acute ischemic stroke, subarachnoid hemorrhage, intracranial atherosclerosis, and vascular cognitive impairment.
Acknowledgments
The authors would like to acknowledge Colby Polonsky, medical illustrator, Georgia Regent’s University for Fig. 1. This work is supported by NIH-NINDS R21 NS081143.
Conflict of Interest
Related posts:
 of Behavioral Deficits in Rodents Following Brain Injury Across Species, Gender, and Experimental Model
of Behavioral Deficits in Rodents Following Brain Injury Across Species, Gender, and Experimental Model
 Data Supporting Glyburide in Ischemic Stroke
Data Supporting Glyburide in Ischemic Stroke
 Ganglia Damage in Experimental Subarachnoid Hemorrhage
Ganglia Damage in Experimental Subarachnoid Hemorrhage
 Pretreatment Fails to Provide Neuroprotection Following a Surgically Induced Brain Injury Rat Model
Pretreatment Fails to Provide Neuroprotection Following a Surgically Induced Brain Injury Rat Model
 IGF-1 Reduced Rat Pup Germinal Matrix Hemorrhage
IGF-1 Reduced Rat Pup Germinal Matrix Hemorrhage
 Injection of Noncellular Cerebrospinal Fluid from Subarachnoid Hemorrhage Patient into Rat Ventricles Leads to Ventricular Enlargement and Periventricular Injury
Injection of Noncellular Cerebrospinal Fluid from Subarachnoid Hemorrhage Patient into Rat Ventricles Leads to Ventricular Enlargement and Periventricular Injury
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


