Overview
Drug treatment is the major form of therapy for a vast majority of children with seizure disorder. Like any other therapy for epilepsy, drug treatment should follow the general principles of management outlined in
Chapter 23. This chapter is concerned with a survey of the AEDs, their metabolism and action in the body, and their indications and mode of practical use.
Most of today’s knowledge about the efficacy and mode of action of AEDs has been obtained in animals. A host of potential anticonvulsant agents has been developed and tested in animal models of experimental seizures, and a number have been found effective in the treatment of human epilepsy. Only a few, however, have withstood extensive clinical trials and the test of time sufficiently well to become part of the therapeutic armamentarium against the epilepsies. During the last decade, many randomized, controlled studies have been performed, primarily in adults but sometimes in children as well. Regularly updated, excellent books dedicated to AEDs testify to the explosion of knowledge in this field (
Levy et al., 2002;
Wyllie, 2001).
Clinical experience has shown that AEDs can control a high proportion of cases of human epilepsy. Moreover, it has demonstrated that certain types of human epileptic seizures respond better to some drugs than to others, even though the correlations between the seizure type and a potentially effective drug are far from perfect (
Eadie and Tyrer, 1989). Various epilepsy syndromes also respond differently to AEDs (
Arzimanoglou, 2002b). Despite the lack of specificity of existing AEDs, clinical practice has shown that a precise diagnosis, when combined with a better understanding of the mechanisms of action of AEDs, permits their more specific and more effective use. Patient characteristics, including age at onset, seizure frequency, electroencephalographic (EEG) data, and the findings of imaging studies, provide diagnostic clues that enable the physician to reach a syndrome diagnosis and to choose the most appropriate AED for each patient.
The
mode of action of AEDs is beyond the scope of this book, so only a few basic notions are offered (
Rogawski, 2002;
Sills and Brodie, 2001). Epileptogenesis does not originate in a single neuron but rather in neuronal pools that have an inherited or an acquired tendency to produce high-frequency bursts of spike discharges (
Najm et al., 2001;
Prince and Connors, 1986). These bursts are then transmitted along axons, and, when a sufficient volume of neural tissue has been activated, a clinical seizure results. Therefore, clinical seizures may be prevented either by lowering the excitability of the pacemaker neuronal pools or by preventing the spread of epileptogenic spike bursts from the pools. Either mechanism may be at work, depending on the drug used, but the prevention of the burst propagation is more significant for most of the conventional anticonvulsants (
Fromm, 1985). This applies especially to primidone (PRM), phenytoin (PHT), carbamazepine (CBZ), and diazepam (
Levy et al., 2002;
Eadie and Tyrer, 1989), whereas some drugs, especially the barbiturates, act mainly by raising the seizure threshold. These presumably act on “epileptic” neurons within the focus.
Anticonvulsant drugs usually bind to specific receptors in the brain.
Receptors are tissue molecules with which drugs form various types of physicochemical bonds through which they exert their therapeutic action. In general, the magnitude of drug effect is related to the number of receptor sites occupied by drug molecules (
Goldstein et al., 1975). The mode of action of an individual drug apparently is closely related to its site of binding (
Levy et al., 2002;
Eadie and Tyrer, 1989). PHT, CBZ, and lamotrigine (LTG), for example, act selectively on sodium channels (
Kuo, 1998), thus preventing repetitive firing of neuronal action potentials, consequently inhibiting the spread of epileptic activity. Other drugs appear to act primarily on synaptic transmission. This action may be presynaptic (e.g., by increasing concentration of γ-aminobutyric acid [GABA] as a result of a decreased catabolism of this compound, as is the case with γ-vinyl-GABA, or by decreasing reuptake within the synaptic cleft), synaptic, or postsynaptic (e.g., binding to specific receptors, thus modifying their excitability). The latter mechanism may involve GABAergic synapses by increasing their activity as a major inhibitory cortical system or excitatory synapses by decreasing the activity of glutamatergic excitatory transmission. Ultimately, all AEDs appear to act on ion channels, although in some cases, the effect is mediated indirectly (
Rogawski, 2002).
Within the limits of current knowledge about the mechanisms of action, most AEDs can be categorized by their mechanisms into several broad categories. Those that act mainly as
voltage-dependent sodium channel blockers include PHT, CBZ, oxcarbazepine (OXC), and LTG. Agents like vigabatrin (VGB) and tiagabine potentiate GABAergic inhibition by enhancing the synaptic availability of GABA. Barbiturates, such as phenobarbital (PB), augment the function of GABAA receptors, and they have additional effects on the calcium and other ion channels. Benzodiazepines, although they enhance only a subset of GABAA receptors, are broad-spectrum agents. Ethosuximide seems to act by affecting T-type calcium channels. Valproate, gabapentin (GPT), felbamate (FBM), topiramate (TPM), zonisamide, and levetiracetam (LEV) appear to have novel mechanisms (or, perhaps, a combination of mechanisms) of action; these may possibly affect the calcium channels and glutamate receptors, as well as conventional targets, including sodium channels and GABA receptor systems (
Rogawski, 2002). Extensive discussions of the mechanisms of action of drugs are available (
Bialer et al., 2002;
Levy et al., 2002;
Eadie and Tyrer, 1989;
Meldrum, 1983a).
Basic Notions on the Pharmacokinetics of Antiepileptic Drugs
Treatment with anticonvulsant agents has long been empirically guided. Recently, the development of clinical pharmacokinetics has enabled the accurate study of the metabolism of drugs through the development of techniques for measuring minute amounts in body fluids. As a result, a large body of new knowledge has been gained about the metabolism of AEDs, their effects relative to the doses given, and the interactions between anticonvulsants and other pharmaceuticals administered to the same patient (
Meldrum, 1983a;
Morselli, 1977).
A number of new concepts have stemmed from pharmacokinetic knowledge, such as the value of monotherapy versus polypharmacy and, in particular, the concept of effective or optimal blood levels of anticonvulsants (
Kutt, 1985;
Richens, 1982).
The concept of
optimal blood levels has often been misinterpreted, to the point that many physicians believe that blood levels outside the therapeutic range are without value and that all prescriptions of an AED should aim for blood levels within that range. The falsity of this interpretation has been amply documented (
Levy et al., 2002;
Perucca, 2000;
Mattson et al., 1992;
Brodie, 1990;
Eadie and Tyrer, 1989;
Holmes, 1987;
Vajda and Aicardi, 1983). The concept of a therapeutic range is statistical, applying only to populations rather than to individual patients. In some patients, the upper or lower limits of individual therapeutic range may lie outside the accepted values, and no effort should be made to achieve therapeutic values in patients who are clinically controlled, even though they are outside the “therapeutic” or optimal range, if they have no clinical evidence of toxicity (
Brodie, 1990;
Vajda and Aicardi, 1983). Blood levels are only one link in the chain of pharmacokinetic processes that occur between ingestion of a drug and that drug then reaching its site of action where it exerts its pharmacodynamic effects. The important factor is, therefore, its concentration at the receptor sites in the central nervous system (CNS).
The concept of an optimal range of a drug applies only if the drug is not irreversibly bound to its receptor and/or if it has active metabolites (
Chadwick, 1988;
Vajda and Aicardi, 1983). Thus, measuring the blood levels of VGB is pointless because this drug is irreversibly bound to GABA transaminase. Likewise, the determination of the blood levels of CBZ or the benzodiazepines is open to criticism because these agents do have active metabolites that may become highly significant clinically when interference from
other pharmaceuticals increases the levels of metabolites (
Agbato et al., 1986).
The value of blood-level estimations is that, for most AEDs, the blood levels are well correlated with the brain levels (
Vajda and Aicardi, 1983;
Sherwin et al., 1973), and the relationship between blood and brain levels is much closer than that between the dosage that was ingested and the blood levels. Blood levels are, therefore, related to therapeutic action. However, exceptions do exist (
Kutt, 1985). In addition, the correlation between blood and brain levels has been verified only on a relatively rough level (e.g., in the white or gray matter), and little research has been conducted on local variations, especially in and around epileptic foci or epileptogenic lesions (
Monaco et al., 1985;
Baron et al., 1983;
Munari et al., 1982a).
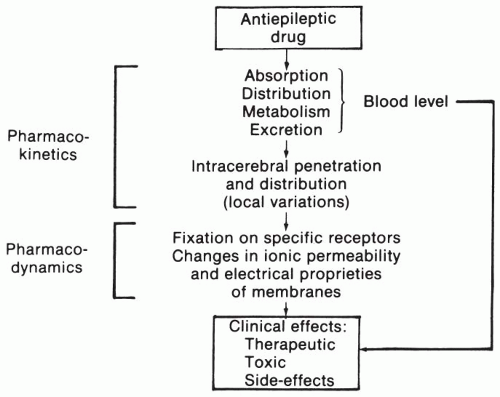
Blood levels of AEDs are the result of a series of processes (
Fig. 24.1). Most anticonvulsants are administered orally and are absorbed rapidly and usually fairly completely (
Table 24.1). Once the drugs are absorbed, they enter the circulation. In the blood, they are variably bound to proteins. The level of protein-bound drugs is in constant equilibrium with that of the free drugs. Only the free fraction can diffuse across the blood-brain barrier to reach the receptor sites. The free fraction of a drug can be altered by various factors (
Bourgeois, 2001). The first is the displacement of the drug from its binding sites by another drug or other chemicals, such as bilirubin (
Morselli, 1977) or free fatty acids. Other factors include pregnancy or age (e.g., infancy or elderly patients). The distribution of drugs varies (
Table 24.1), with the apparent volume of distribution depending particularly on the lipid solubility of particular agents. Some AEDs, such as the benzodiazepines, have volumes of distribution that are greater than the total body water, thus indicating binding to tissue constituents, active transport into cells, or accumulation in storage areas. Others have volumes of distribution equal to or approaching that of total body water, while valproic acid (VPA) has a volume of distribution close to that of the extracellular water.
Anticonvulsants leave the blood by the following two mechanisms: (a) biotransformation to inactive or, less commonly, active metabolites and (b) excretion through the kidney, intestine, sweat, or tears. “Biotransformation” is an enzymatic process resulting mainly from hepatic microsomal activity. This, in turn, depends on the genetic makeup and gender of the patient, on the one hand, and on a number of extraneous factors such as diet and interaction with other drugs that induce the synthesis of microsomal drug-metabolizing enzymes, on the other. Interference also may result from inhibition of the metabolism of one drug by another through various mechanisms (
Kutt, 1985;
Perucca and Richens, 1980). The excretion of drug also can be altered by such factors as urinary pH level, urine flow, or the glomerular filtration rate.
The apparent
plasma half-life of a drug is defined as the time it takes for the plasma level to decline to 50% of its previous value. The decline usually occurs exponentially. The average plasma half-lives of the main anticonvulsant drugs are indicated in
Table 24.1. Considerable variations exist, and these values are
only a general indication. Moreover, the half-life can be influenced by a number of factors affecting excretion and/or biotransformation. For example, the half-life of many drugs tends to decrease, often considerably, as a result of the microsomal enzymatic induction that is provoked by the administration of the drug itself (autoinduction) or by that of another drug (
Perucca and Levy, 2002;
Bourgeois, 1988,
1992;
Eadie and Tyrer, 1989;
Bourgeois and Wad, 1988).
The time course of drug concentrations in different fluids or tissues can be fitted to mathematical models that are used for interpreting the plasma concentration pattern obtained in a given patient. Many pharmacokinetic processes, especially excretion and biotransformation of a drug, are said to be first order. In that situation, the elimination process varies directly with the plasma concentration of the drug, and a linear relationship between the dosage and serum levels is observed. When the elimination is independent of the concentration or dose that is administered, the process is said to be “zero order.” Many drugs convert from first-order to zero-order kinetics when their concentration becomes sufficiently high to saturate their enzyme or transport mechanism. That change is clinically significant when PHT is being used because the saturation point is within the average therapeutic concentration range. When saturation is attained, minimal incremental increases in dose produce considerable increases in plasma levels, with attendant toxicity.
As has already been indicated,
variability is the rule rather than the exception for most pharmacokinetic processes. Part of this variability is intrinsically determined. For example, the following two populations exist with regard to the speed of hydroxylation of PHT: slow and fast hydroxylators (
Eadie and Tyrer, 1989;
Richens, 1982). A considerable part of the variation results from extraneous processes, among which interactions, especially drug interactions, are important (
Kutt, 1984). Interactions can take place at any step from absorption to interaction with specific receptors. Among antimicrobial agents, chloramphenicol may cause the accumulation of PHT and PB,
and isoniazid may cause PHT, CBZ, and PRM to accrue. Erythromycin may cause the buildup of CBZ. Among antiulcer agents, antacids may reduce PHT concentration, whereas cimetidine may cause the accumulation of PHT, CBZ, and diazepam. Salicylates displace strongly bound drugs such as PHT, diazepam, or VPA from the binding sites in plasma proteins, which may lead to some decline of the total plasma level and a corresponding increase in the unbound drug percentage. However, only a few of these interactions are clinically significant, necessitating the adjustment of drug dosages. In children, one important example is the toxicity produced by the coadministration of CBZ and some antibiotics, especially triacetyloleandomycin (
Mesdjian et al., 1980) and erythromycin (
Goulden et al., 1986), which considerably increase the levels of CBZ.
The timing of ingestion of valproate relative to meals, as well as the composition of the meals, markedly influences the absorption of that drug (
Loiseau et al., 1982;
Lévy et al., 1981) and the time at which peak and trough plasma levels are achieved. The galenic form of drugs can also significantly influence their bioavailability and rate of absorption (
Issakainen and Bourgeois, 1987;
Wyllie et al., 1987b;
Hodge et al., 1986), and poor preservation of drugs may also be responsible for variations. Thus, storing PHT and CBZ in a hot and humid place can reduce their bioavailability by up to 50% (
Cloyd, 1991). Extremely rare cases of malabsorption of AEDs have been recorded (
Gilman et al., 1988). These may be important, especially with short half-life drugs, for determining the optimal time for sampling blood for a plasma level determination. Following a gastrostomy, one of the authors’ patients who was on slow-release sodium valproate experienced a relapse of seizures. The competition of drugs for protein-binding sites may produce significant interaction when both of the agents that are used are strongly protein bound. Valproate can displace PHT from its albumin sites, with resultant increases in free fraction. At that point, the determination of the PHT blood levels, in which both free and protein-bound fractions are measured, does not give a true picture of the pharmacokinetic situation because the active (free) fraction is abnormally high relative to the total level. Toxicity may result if the PHT dose is increased without taking that phenomenon into account (
Richens, 1982;
Perucca and Richens, 1980). At a second stage, the increases in the free fraction augment the clearance of PHT, thus reestablishing the original equilibrium, often with a lower total level of the pharmaceutical in the blood (
Perucca and Richens, 1980).
The determination of blood levels of free drugs rather than the total blood level (free plus protein-bound fractions) may obviate some of these difficulties (
Herngren et al., 1991;
Gianelli et al., 1988;
Agbato et al., 1986;
Lévy and Schmidt, 1985). However, the technique for such a determination is more complicated (
Pacifici and Viani, 1992), so a measurement of the free levels of AEDs has not supplanted total level determination, except in special circumstances such as pregnancy, hypoalbuminemia, and hepatic or renal failure (
Lévy and Schmidt, 1985). An indirect approach to the determination of free drug level is by using biological fluids other than blood for determination. Tears and, in particular, saliva levels have been studied and are used in several places (
Drobitch and Svensson, 1992;
Knott, 1983;
Kristensen et al., 1983). Although these are relatively protein-free fluids, their concentration of AEDs may vary with the secretion rate and other factors, and sampling may be more difficult (
Bäckman et al., 1987).
The induction of microsomal drug-metabolizing enzymes can result in drug interference. The clinical significance of this type of interference is usually limited (
Perucca and Levy, 2002;
Patsalos and Lascelles, 1982;
Perucca and Richens, 1980). However, CBZ may reduce the blood levels of PHT and vice versa, partly through that type of interaction (
Bourgeois, 1988;
Browne et al., 1988b), and many interactions of at least some clinical significance are known (
Pippenger, 1987;
Pisani et al., 1987;
Patsalos and Lascelles, 1982). The failure of oral contraceptives has been attributed to anticonvulsants in some patients. Probably, the most predictable interaction necessitating dosage adjustment is the accumulation of PB that is caused by valproate (
Kutt, 1984). The most significant interactions result from the inhibition of the metabolism of one drug by another. This occurs with the coadministration of PB and valproate, in which valproate produces an average increase of 30% in the blood levels of PB (
Scheyer, 2002). PRM concentrations, as well as the derived PB concentrations, increase when valproate is added. However, large variations ranging from 0% to more than 100% can be observed in individual patients.
Valproate blocks the glucuronidation of LTG and results in the increased concentration of LTG, so the dose used should be cut to about half when such a combination is prescribed. On the other hand, the metabolism of LTG, like that of many other drugs, is accelerated with combined use with other inducing anticonvulsants, so the dose may need to be increased when these are used together.
Pharmacokinetic interactions can be evaluated rather easily by measuring changes in blood concentrations. However, measuring the so-called
pharmacodynamic interactions, which manifest by a change in the pharmacologic response in the absence of changes in blood levels, objectively has proven much more difficult (
Perucca and Levy, 2002). Pharmacodynamic interactions may be expressed negatively (additive neurotoxicity), usually leading to the appearance of CNS side effects in patients on polytherapy, even when the doses and blood levels of individual AEDs are in the low range (
Besag et al., 1998). The combination of drugs on an empirical basis also produces some favorable interactions, providing a superior therapeutic index. Clinical evidence is convincing for the combination of valproate with ethosuximide for the control of refractory absence seizures (
Rowan et al., 1983) and for valproate with LTG in patients with generalized epilepsies (
Arzimanoglou et al., 2001;
Brodie and Yuen, 1997;
Panayiotopoulos et al., 1993) or partial seizures (
Pisani et al., 1999). Potential advantages have been reported for other combinations, but the evidence is mostly anecdotal and interindividual variation can be considerable (
Perucca and Levy, 2002).
In children, an additional and major source of variability is the
age of the patient. Age influences all aspects of drug metabolism (
Dodson, 1983,
1987b;
Morselli et al., 1983). The absorption of AEDs, except CBZ, is usually faster in infants and young children than in adults (
Dodson, 1987a). The half-lives of most anticonvulsants are quite prolonged in newborn babies during the first 1 to 3 weeks of life, and they shorten dramatically thereafter over the first 2 or 3 months of life. High metabolic levels are maintained during the first years of life. At 10 to 15 years, a marked metabolic slowing to adult values often occurs (
Dodson, 1987b). This slowing may be progressive, or, on some occasions, it may be quite sudden (
Morselli, 1977). The evolution of drug metabolism explains why intoxication is common in neonates receiving repeated doses of anticonvulsants over the first days of life, whereas, a few days or weeks later, maintaining adequate therapeutic levels may be difficult or even impossible (
Arzimanoglou and Aicardi, 2001). This also accounts for the higher doses per unit of weight or surface area that are required in children compared with adults. Finally, the slowing of metabolism at the time of adolescence may account for the relative frequency of overdosage at that period of life despite the increase in body weight.
Other sources of variability in the doses required and the blood levels necessary to control the seizures depend on the individual variations in the severity of epilepsy, pharmacologic tolerance, and other unknown causes.
Significant changes can occur also with stress (e.g., trauma or surgery), which can increase the metabolism of drugs, such as valproate or CBZ, consequently diminishing their blood level; however, the effectiveness is not necessarily diminished when CBZ is used because the increased level of the epoxide is also active and potentially toxic (
Cloyd, 1991). Febrile illnesses can also alter PHT clearance (
Leppik et al., 1986).
Reciprocally antiepileptic agents, many of which are strong microsomal inducers, can alter the metabolism and efficacy of simultaneously used drugs (
Perucca and Levy, 2002). For example, one important interference is seen in patients who receive corticosteroids to avoid rejection of a transplant, in whom the acceleration of the catabolism of steroids may lead to acute rejection (
Pentella et al., 1982). In such cases, the use of noninducing drugs, such as valproate, LTG, GPT, or the benzodiazepines, is advised. The same type of problem may present with the combined use of warfarin and some anticonvulsant drugs, including CBZ, PB, and PHT, which induce warfarin (
Levy et al., 2002). The interference of anticonvulsants with the metabolism of vitamin D has resulted in decreases in calcium fixation, thereby repeatedly causing the occurrence of osteomalacia or rickets (
Chung and Ahn, 1994;
Offerman et al., 1979). This does, however, not appear to be of major clinical importance in ambulatory children for whom no increase in vitamin D administration is recommended (
Ala-Houala et al., 1986). In institutionalized patients, the routine administration of vitamin D supplementation may be indicated.
The considerable magnitude of individual variations from all sources is one of the reasons that rigid schemes or protocols cannot be established for the treatment of the epilepsies of childhood.
PB, PHT, and CBZ are potent inducers of drug metabolism, and their administration is usually associated with decreases in levels of CBZ (heteroinduction and autoinduction), cyclosporine, oral contraceptives, and corticosteroids. OXC, TPM, and FBM have a narrower spectrum of enzyme-inducing activity, but they have also been found to reduce the blood levels of steroid and oral contraceptives (
Perucca and Levy, 2002).