CHAPTER 48 Meningiomas and Neurofibromatosis Type 2
INTRODUCTION
The neurofibromatoses consist of two different, dominantly inherited disorders, neurofibromatosis type 1 (NF1) and neurofibromatosis type 2 (NF2). Although NF2 was first described in 1822 by the Scottish surgeon James Wishart and NF1 was fully delineated in the late 19th century by von Recklinghausen, until the late 1980s these two different entities were thought to represent different presentations of a single disease.1–3 After the localization of the NF1 and NF2 genes to chromosome 17 and 22, respectively, these two conditions were clearly established as separate entities.4–6 Table 48-1 demonstrates the different features of these two distinct conditions.
TABLE 48-1 Differences between neurofibromatosis type 1 and type 2
| NF1 | NF2 | |
|---|---|---|
| Inheritance | Autosomal dominant | Autosomal dominant |
| Spontaneous mutations | 50% | 50% |
| Incidence | 1:3000 | 1:40,000 |
| Chromosome | 17q11.2 | 22q12.2 |
| Gene product | Neurofibromin | Merlin |
| Typical presentation | Café-au-lait macules during infancy or early childhood | Hearing loss or vestibular dysfunction in young adulthood or cataracts |
| Nerve sheath tumors | Neurofibroma, plexiform neurofibroma, MPNSTs | Schwannoma |
| Intracranial tumors | Optic pathway gliomas, other astrocytomas/gliomas | Vestibular schwannomas, meningiomas |
| Spinal tumors | Nodular plexiform neurofibromas (roots) | Schwannomas (roots), ependymomas (intramedullary) |
| Cutaneous features | Café-au-lait macules, axillary/inguinal freckling, cutaneous neurofibromas, subcutaneous neurofibromas | Cutaneous schwannomas |
| Cognitive | LD and ADHD common, IQ mildly decreased | Normal |
| Ophthalmologic | Lish nodules, congenital glaucoma | Juvenile subcapsular lenticular opacities, cataracts, corneal scarring, retinal hamartomas |
MPNST, malignant peripheral nerve sheath tumor; LD, learning disability; ADHD, attention deficit and hyperactivity disorder
Adapted from ref. 16.
NF2, also known as “central neurofibromatosis” or “bilateral acoustic neurofibromatosis,” is an autosomal dominant disease caused by mutations of the NF2 tumor suppressor gene.7,8 Accordingly, the risk to develop this disease for any offspring of an affected parent is 50%.4 Fifty percent of the cases have no family history and represent sporadic gene mutations.9,10 The birth incidence of the disease based on a study on the UK population is estimated between 33,000 and 40,000 with a prevalence of 1 in 210,000. The annual incidence rate is 1 per 2,355,000 (newly diagnosed symptomatic patients), which represents about one new case per year.9 The difference between the birth incidence and diagnostic prevalence is the result of the fact that many patients do not develop the disease until the third decade or later, and many others die before that time.1,2
Using genetic analyses of large pedigrees, the NF2 gene has been localized to the long arm of chromosome 22. The disease results from inactivating mutations of a known tumor suppressor gene. The gene is comprised of 110 kb of genomic DNA and encodes a 595-amino-acid protein termed merlin or schwannomin.2,9,11–13 Merlin is structurally related to the moesin/ezrin/radixin proteins, which link the actin cytoskeleton to cell surface glycoproteins that control growth and cellular remodeling.2,14 Merlin is widely expressed in Schwann cells, meningeal cells, peripheral nerves, and the lens. There is evidence that the main function of the NF2 gene is tumor suppression and regulation of proliferation of Schwann cells and leptomeningeal cells.2 Inactivating mutations and the loss of merlin expression were also detected in almost all sporadic schwannomas, in 50% to 70% of sporadic meningiomas, gastrointestinal stromal tumors, malignant mesothelioma, and sporadic ependymomas.15,16
CLINICAL DIAGNOSIS OF NF2
In 1987, the National Institutes of Health (NIH) Consensus Development Conference on Neurofibromatosis established clinical diagnostic guidelines to differentiate NF2 from NF1.17 These criteria diagnose NF2 in patients with bilateral vestibular schwannomas and in those with a first-degree relative with NF2 and either a unilateral vestibular schwannoma or at least two other characteristic features of NF2 (meningioma, schwannoma, glioma, neurofibroma, juvenile posterior subcapsular lenticular opacity). However, half of all patients with NF2 do not have a family history of the disease, and patients can present with other features of the disease long before the appearance of vestibular schwannomas.10,18–20 Owing to the drawbacks in the diagnosis of NF2 in patients without bilateral vestibular schwannomas and without a positive family history, these criteria were revised multiple times. A second NIH Consensus Conference in 1991, the Manchester group in 1992, and a group organized by the National Neurofibromatosis Foundation (NNFF) in 1997 revised these criteria.7 Table 48-2 demonstrates the latest diagnostic criteria for NF2. Baser and colleagues have examined the diagnostic efficiency of the four sets of criteria in patients who do not have bilateral vestibular schwannomas but who do have other signs of NF2 and concluded that although none of the sets is satisfactory for diagnosing patients without bilateral vestibular schwannomas, the Manchester criteria are the most sensitive.7
TABLE 48-2 Evolution of clinical diagnostic criteria for NF2
| 1997 NFFF criteria |
|---|
| 1991 NIH criteria |
|---|
| 1987 NIH criteria |
|---|
* In the Manchester criteria, “any two of” refers to two individual tumors or cataract, whereas in the other sets of criteria, it refers to two tumor types or cataract.
Adapted from ref. 7.
CLINICAL MANIFESTATIONS
The hallmark of NF2 is bilateral vestibular schwannomas (Fig. 48-1). The penetrance of vestibular schwannomas in families with the NF2 gene is greater than 95%9 and this pathognomonic finding is virtually present in 95% of adult patients.10,20,21 Besides these tumors, the clinical presentation of the disease is quite heterogeneous. One can now classify patients into two main groups, severe and mild phenotypes, based on differing clinical course, age at onset, and the presence of other cranial and spinal tumors.19 The severity of the disease is mainly associated with type of mutation. Studies of genotype and phenotype correlations in NF2 have demonstrated that, in general, constitutional nonsense and frameshift mutations in the NF2 gene are associated with severe disease, while missense mutations, in-frame deletions, large deletions, and somatic mosaicism are associated with mild illness.10,19,22–24
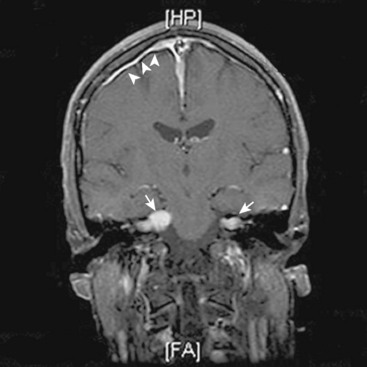
Although different authors describe cutoff points for severe and mild disease with different numbers, the mild phenotype, which is referred to as the “Gardner” form, is characterized by late onset of bilateral vestibular schwannomas (usually after the second decade of life) with limited numbers of additional tumors of the cerebrum and spine. On the other hand, the severe phenotype, which is referred as the “Wishart” form, is associated with numerous tumors of the cranium and the spine with early-onset vestibular schwannomas.9,19,25 The severe phenotype usually presents with a tumor other than a vestibular schwannoma in early childhood and is associated with a high morbidity and mortality.1,25
The average age at onset of symptoms in individuals with NF2 is 18 to 24 years; however, they can present as early as 2 years and as late as 70 years.9 The average age of mortality is 36 years.22 There is no preponderance for sex and race.
Unilateral hearing loss, symptoms related to skin tumors, focal weakness due to spinal tumors or neuropathies, tinnitus, and imbalance are the most common symptoms in affected adults. Eleven percent of the cases are asymptomatic and detected on screening because of a positive family history.1,9,19,25
Ophthalmologic features are common in NF2. About 30% of the cases have decreased visual acuity in one or both eyes. Between 60% and 80% of the patients have posterior subcapsular lenticular opacities. Retinal hamartoma, epiretinal membranes, optic nerve meningioma, optic disc glioma, intraocular schwannoma, and neurotrophic keratopathy are other manifestations of NF2. Lisch nodules were not found to be a feature of the disease.1,3,10,19,26,27
Cutaneous features in NF2 are much rarer than in NF1. Although 70% of the patients have skin tumors, the number of these tumors does not exceed 10 in most of the cases. Café au lait spots do occur in about 40% of NF2 patients, but again the size and number of these lesions are far less than in NF1. Fewer than 2% of NF2 patients harbor more than six café au lait spots, which may pose some diagnostic confusion between NF1 and NF2.12 Axillary and groin freckling is not a characteristic of the disease. Skin tumors occur in about 70% of the patients and the most common type of these tumors is intracutaneous, slightly raised plaque-like lesions. Schwannoma is the main histopathologic diagnosis, but occasionally neurofibromas occur. The back, chest, arms, and face are the most common locations.1,4,10,19,28
Although rare, recent studies have highlighted peripheral nerve involvement that cannot be attributed to tumors in NF2 patients.21,29 Evans and colleagues1 have reported peripheral nerve lesions unrelated to tumors in 6% of NF2 cases. Sperfeld and colleagues investigated definite NF2 patients and found clinical and electrophysiologic signs related to peripheral neuropathy in the majority of the cases.30
The presentation of the disease in childhood is quite different that that in adults. Although NF2 is recognized as an adult-onset disease, many patients are found to have had some signs of the disease in early childhood.18,31 Unlike the adulthood presentation, vestibular schwannoma–related symptoms occur in as few as 15% to 30% of patients as initial symptoms. Patients may present with meningiomas and spinal tumors long before vestibular schwannomas. Children present more frequently with neurologic symptoms primarily related to spinal cord compression and visual involvement.18 Cataracts can be present in the neonatal period and can affect vision in early life. In addition, there is a tendency to mononeuropathy, particularly affecting the facial nerve.1 Some children may present with oculomotor palsy or a foot or hand drop.1,18,20,31 NF2 patients who present in childhood are likely to have a more severe disease course with multiple tumors.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


