Fig. 7.1
Low power view showed the small round blue cell component of the mesenchymal chondrosarcoma attached to the dura mater (H&E, ×20)
Clinical Presentation
The clinical presentation of intracranial mesenchymal chondrosarcoma is similar to those of other mass lesions within the CNS. Symptoms and signs are dependent on the location of the tumour, and frequently reflected the compressive effects of involved neurological structures, with examples including cranial nerve palsies in tumours involving the base of skull. Most patients have a tendency to present with headaches and other symptoms related to increased intracranial pressure (Scheithauer and Rubinstein 1978), and isolated cases may only be detected after the discovery of metastatic disease.
Radiological Features
Imaging of intracranial mesenchymal chondrosarcoma show inconsistent features, however, the majority of cases are hypointense to normointense on T1-weighted MRI, with strong enhancement after administration of gadolinium (Huang et al. 2004). It can be extremely hypervascular on angiography, and embolisation may be required prior to surgery. Due to its common involvement of the meninges and strong enhancement on MRI, these tumours may resemble malignant meningioma or haemangiopericytoma on radiological imaging (Chen et al. 2004).
Histopathology
Pathological examination of both extraosseous and osseous examples shows the same characteristic biphasic appearance (Scheithauer and Rubinstein 1978). The two distinct elements include islands of hyaline cartilage and a proliferation of undifferentiated small round cells (Fig. 7.2). The cartilaginous component is well differentiated and may appear as benign hyaline cartilage, low-grade chondrosarcoma, and rarely, as an intermediate-grade chondrosarcoma (Unni et al. 2005). The undifferentiated areas show sheets or alveolar arrangements of small round cells with relatively uniform nuclei, dense chromatin and sparse cytoplasm (Fletcher et al. 2002; Unni et al. 2005; Louis et al. 2007). Staghorn vascular spaces are commonly seen. The two characteristic components have a tendency to show an abrupt transition, however, occasional cases may show a gradual merging of the two elements. More importantly, the proportion of each element is highly variable and diagnostic problems arise when a limited biopsy only shows one component (Bingaman et al. 2000; Lin et al. 2012).
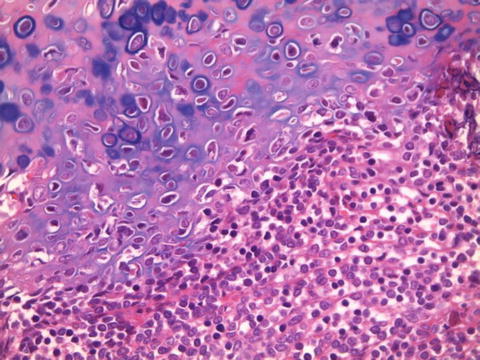
Fig. 7.2
Section showed the characteristic biphasic histological appearance, with islands of hyaline cartilage juxtaposed against a small round blue cell population (H&E, ×200)
Histochemical and Immunoperoxidase Studies
Histochemical and immunoperoxidase studies of mesenchymal chondrosarcoma are not specifically helpful in distinguishing it from other differential diagnoses. The small cells are glycogen rich (Burger and Scheithauer 2007), and stain positively for CD99, vimentin and Leu7. The chondroid cells are positive for S100 (Fletcher et al. 2002). Sox9, a master regulator of chondrogenesis, has been shown in a study by Wehrli et al. (2003) to reliably differentiate mesenchymal chondrosarcoma from other small round blue cell tumours, based on the hypothesis that both small cell and cartilaginous components are derived from primitive chondroprogenitor cells. In their study, 21 of 22 cases of mesenchymal chondrosarcoma showed positive nuclear staining in both components, and the remaining 73 small round blue cell tumours (including rhabdomyosarcoma, neuroblastoma, Ewing sarcoma/PNET, lymphoma, small cell carcinoma, merkel cell carcinoma, small cell osteosarcoma, extraskeletal myxoid chondrosarcoma and small cell desmoplastic tumour) displayed negative staining. Hence, Sox9 may be an useful ancillary stain for diagnosing mesenchymal chondrosarcoma when an undifferentiated small round cell component is present with a lack of chondroid elements.
Ancillary Tests
Electron microscopy of the undifferentiated small cells show large nuclei and little organelles, similar to primitive mesenchymal cells, and the cartilaginous areas show a usual chondrocyte appearance (Fletcher et al. 2002; Unni et al. 2005). No specific molecular aberration has been identified, although an identical Robertsonian translocation involving chromosomes 13 and 21 [der(13;21)(q10;q10)] has been detected in two cases (Fletcher et al. 2002; Unni et al. 2005).
Diagnostic Complexities
As mentioned previously, the proportion of the two components is highly unpredictable and the predominance of either component may result in diagnostic difficulties. The dominance of cartilaginous component may lead to a misdiagnosis of a pure chondroid lesion, and the sole presence of the small round cell component may result in the erroneous diagnosis of other small round cell tumours. Even if the tumour contained both elements in significant amounts, diagnostic dilemma may arise if only one element is sampled in a limited biopsy or debulking procedure.
Furthermore, a recent case report by Lin et al. (2012) highlighted a diagnostic pitfall in the diagnosis of mesenchymal chondrosarcoma arising from the tentorium cerebelli of a 21 year old woman. Histological examination of the tissue obtained at the initial debulking procedure demonstrated a dural-based pure small round cell population with hyperchromatic ovoid cells, finely to coarsely granular chromatin, inconspicuous nucleoli and scanty cytoplasm. The cells were Periodic acid Schiff (PAS) positive and diastase sensitive, and immunohistochemically showed focal strong membranous immunoreactivity with CD99, and negative staining for epithelial markers. The diagnosis of Ewing sarcoma/primitive neuroectodermal tumour (PNET) was made and the patient underwent chemotherapy and radiotherapy according to a PNET protocol. However, the tissue obtained during a definitive complete macroscopic removal several months after the initial procedure showed prominent hypercellular lobules of atypical chondroid cells, prompting the re-diagnosis as a mesenchymal chondrosarcoma. This case highlighted diagnostic complexities of mesenchymal chondrosarcomas in an intracranial location where limited biopsy material is often obtained, and potential misdiagnosis if only one component is sampled. The authors also raised a second hypothesis of post-chemotherapy cellular maturation in an attempt to explain the differences in histological appearances seen in the tissue obtained from the first and second procedure. Post-chemotherapy cellular maturation is a well known phenomenon described in a range of pediatric tumours, including pediatric sarcomas and embryonal tumours, Wilms’ tumour and germ cell tumours (McCartney et al. 1984; Omar et al. 1986; Coffin et al. 2005; Smith et al. 2002; Nozza et al. 2010). This phenomenon is thought to be a secondary event to anti-cancer agents selectively destroying immature and more anaplastic clones, and hence, offering relatively benign and mature cells a survival advantage (McCartney et al. 1984; Omar et al. 1986). The post-chemotherapy cytomaturation phenomenon may potentially explain the marked differentiation of the cartilaginous component in this case.
Related posts:
 Oligodendroglial Tumors: Intra-arterial Chemotherapy
The Concept of a Preniche for Localization of Future Metastases
Differentiating Choroid Plexus Tumors from Metastatic Carcinomas: Use of Inwardly Rectifying K+ Channel KIR7.1 and Excitatory Amino Acid Transporter-1
Oligodendroglial Tumors: Intra-arterial Chemotherapy
The Concept of a Preniche for Localization of Future Metastases
Differentiating Choroid Plexus Tumors from Metastatic Carcinomas: Use of Inwardly Rectifying K+ Channel KIR7.1 and Excitatory Amino Acid Transporter-1
 Use of Mobile Phones and Brain Cancer Risk in Children?
Use of Mobile Phones and Brain Cancer Risk in Children?
 Metastatic Oligodendroglioma: Diagnosis with Fine-Needle Aspiration Cytology
Metastatic Oligodendroglioma: Diagnosis with Fine-Needle Aspiration Cytology
 Lipoma: An Overview
Lipoma: An Overview
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


