Symptoms
Metabolic defect
Clinical manifestations
Myopathy, stress intolerance
Mitochondrial disorder
Electron transport chain defects
Lysosomal disorder
Glycogen storage disease type II, Danon’s disease
Defect in purine metabolism
Myoadenylate deaminase deficiency
Neuropathy
Lysosomal disorder
Fabry’s disease, Krabbe’s disease, metachromatic leukodystrophy
Peroxisomal disorder
Refsum’s disease, adrenoleukodystrophy/adrenomyeloneuropathy
Mitochondrial disorder
Electron transport chain defects
Defect in cholesterol metabolism
Cerebrotendinous xanthomatosis
Defect in heme biosynthesis
Porphyria
Spinal muscular atrophy
Lysosomal disorder
GM2 gangliosidosis
Spasticity
Mitochondrial disorder
Electron transport chain defects
Lysosomal disorder
GM1 gangliosidosis, GM2 gangliosidosis, Gaucher’s disease type 3, Krabbe’s disease, metachromatic leukodystrophy, Salla disease
Peroxisomal disorder
Refsum’s disease, adrenoleukodystrophy/adrenomyeloneuropathy
Defect in cholesterol metabolism
Cerebrotendinous xanthomatosis
Defect in urea cycle
Arginase deficiency
Progressive ataxia
Mitochondrial disorder
Pyruvate dehydrogenase deficiency, electron transport chain defects
Lysosomal disorder
GM2 gangliosidosis, metachromatic leukodystrophy, Niemann-Pick disease type C, sialidosis, Salla disease
Peroxisomal disorder
Refsum’s disease, adrenoleukodystrophy/adrenomyeloneuropathy
Defect in cholesterol metabolism
Cerebrotendinous xanthomatosis
Defect in copper metabolism
Wilson’s disease
Defect in lipid metabolism
Abetalipoproteinemia
Mitochondrial disorder
Electron transport chain defects
Lysosomal disorder
GM1 gangliosidosis, GM2 gangliosidosis (Tay-Sachs disease, Sandhoff’s disease), metachromatic leukodystrophy, Niemann–Pick disease type C, neuronal ceroid lipofuscinosis (Spielmeyer–Vogt disease, Kufs’ disease)
Peroxisomal disorder
Adrenoleukodystrophy
Defect in cholesterol metabolism
Cerebrotendinous xanthomatosis
Defect in copper metabolism
Wilson’s disease
Defect in purine metabolism
Lesch-Nyhan syndrome
Organic acid disorder
Glutaric aciduria type I
Leukodystrophy/leukoencephalopathy (specific patterns of distribution)
Mitochondrial disorder
Lysosomal disorder
Peroxisomal disorder
Defect in cholesterol metabolism
Organic acid disorder
Electron transport chain defects
Krabbe’s disease, metachromatic leukodystrophy
Adrenoleukodystrophy
Cerebrotendinous xanthomatosis
Canavan’s disease
(Myoclonic) epilepsy
Mitochondrial disorder
Lysosomal disorder
Pyruvate dehydrogenase deficiency, electron transport chain defects
Sialidosis, neuronal ceroid lipofuscinosis (Spielmeyer-Vogt disease, Kufs’ disease)
Behavioral abnormalities, psychosis, dementia
Mitochondrial disorder
Lysosomal disorder
Electron transport chain defects
Fabry’s disease, GM2 gangliosidosis (Tay–Sachs disease, Sandhoff’s disease), Gaucher’s disease type 3, metachromatic leukodystrophy, Niemann-Pick disease type C, neuronal ceroid lipofuscinosis (Spielmeyer-Vogt disease, Kufs’ disease)
Peroxisomal disorder
Adrenoleukodystrophy
Defect in cholesterol metabolism
Cerebrotendinous xanthomatosis
Defect in copper metabolism
Wilson’s disease
Defect in amino acid metabolism
Homocystinuria
Defect in urea cycle
Ornithine transcarbamylase deficiency
Loss of vision
Mitochondrial disorder
Electron transport chain defects
Lysosomal disorder
Neuronal ceroid lipofuscinosis (Spielmeyer-Vogt disease), sialidosis
Peroxisomal disorder
Refsum’s disease
Defect in urea cycle
Ornithine aminotransferase deficiency
Ophthalmoplegia
Mitochondrial disorder
Electron transport chain defects
Lysosomal disorder
Gaucher’s disease type 3, Niemann-Pick disease type C
Stroke, stroke-like episodes
Mitochondrial disorder
Electron transport chain defects
Lysosomal disorder
Fabry’s disease
Defect in amino acid metabolism
Homocystinuria, methylene tetrahydrofolate reductase deficiency
Defect in urea cycle
Ornithine transcarbamylase deficiency
Recurrent attacks of ataxia
Mitochondrial disorder
Pyruvate dehydrogenase deficiency, electron transport chain defects
Defect in amino acid metabolism
Branched-chain organic aciduria, methylene tetrahydrofolate reductase deficiency
Defect in urea cycle
Ornithine transcarbamylase deficiency
Recurrent psychiatric symptoms
Defect in amino acid metabolism
Methylene tetrahydrofolate reductase deficiency
Organic acid disorder
Branched-chain organic acid disorder
Defect in urea cycle
Ornithine transcarbamylase deficiency
Defect in heme biosynthesis
Acute porphyria
Mitochondrial Diseases
Definition of Mitochondrial Diseases
The term “mitochondrial diseases” does not apply to all disorders of metabolism in mitochondria: it refers only to impairments of the energy metabolism, particularly those of the electron transport chain (respiratory chain), and in part also to disturbances of the immediate pyruvate metabolism and the citrate cycle.
Epidemiology
The estimated prevalence of mitochondrial diseases is 15 per 100 000 head of population.
Physiology and Etiology
Mitochondria are cellular organelles surrounded by a double membrane; inside these organelles, diverse metabolic processes take place (Fig. 17.1):
• Dehydrogenation and decarboxylation of pyruvate to acetyl CoA.
• The subsequent citrate cycle.
• Oxidative phosphorylation in the electron transport chain.
• β-Oxidation of fatty acids.
• Parts of the urea cycle.
The electron transport chain, in which the reoxidation of coenzymes is coupled to the synthesis of adenosine triphosphate (ATP)—the central energy source of the cell—consists of five enzyme complexes:
• Complex I: ubiquinone reductase.
• Complex II: succinate dehydrogenase.
• Complex III: cytochrome c oxidoreductase.
• Complex IV: cytochrome c oxidase (COX).
• Complex V: ATP synthase.
Mitochondria are inherited maternally and contain several copies of their own mitochondrial DNA (mtDNA). The mitochondrial DNA consists of a circular double strand of 16 569 base pairs; it codes for 13 polypeptides of the electron transport chain (mit genes) and also for two ribosomal RNAs and 22 transfer RNAs (syn genes). The majority of mitochondrial proteins, however, are encoded by nuclear DNA (nDNA) and are imported into the mitochondria after synthesis. Gene products of nDNA are also essential for maintaining and replicating the mtDNA. Normally, an organism contains only one mtDNA species. When a mutation occurs, a cell often contains both mutant mtDNA and wildtype mtDNA (heteroplasmy), with both being randomly distributed during cell division. As a result, one cell may receive only mutant DNA and another only wild-type DNA (segregation). Cell functions are only affected when the proportion of mutant mtDNA within a cell exceeds a critical threshold level. This explains, in part, the different manifestations of mtDNA mutations within a family.
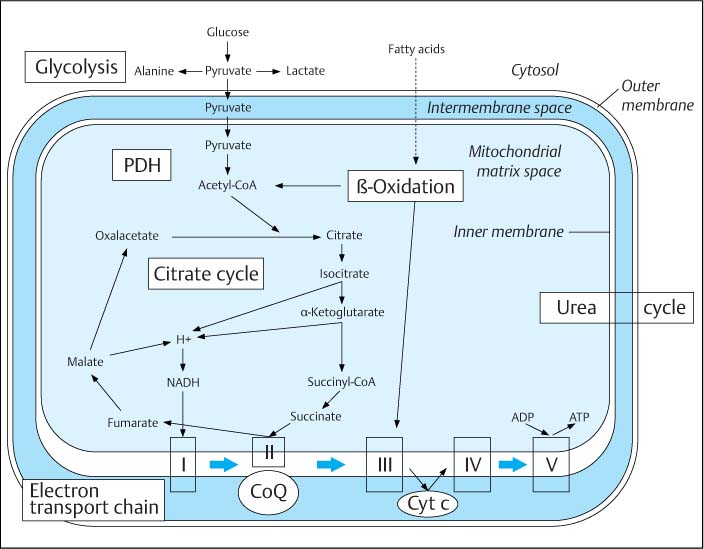
Fig. 17.1 Mitochondrial metabolism.
PDH, pyruvate dehydrogenase Electron transport chain (respiratory chain):
Complex I | —ubiquinone reductase |
Complex II | —succinate CO oxidoreductase |
Complex III | —cytochrome c oxidoreductase |
Complex IV | —cytochrome c oxidase (COX) |
Complex V | —ATP synthase |
CoQ | —coenzyme Q |
Cyt c | —cytochrome c |
Clinical Features
Defects in the mitochondrial electron transport chain may manifest in any organ and at any age during life. They may show very variable patterns of inheritance, and they may be chronic or rapidly progressive. Table 17.2 presents an overview of possible symptoms of an electron transport chain defect. The cardinal symptoms of pyruvate dehydrogenase (PDH) deficiency include developmental delay, epilepsy, ataxia, and progressive encephalopathy.
If there are two symptoms that cannot be explained otherwise, and particularly if they belong to different organ systems, the possibility of a mitochondrial disorder should be considered.
However, mitochondrial disease can also manifest as pure myopathy. Typical constellations of clinical features have been grouped together and designated as syndromes (Table 17.3). Whereas mtDNA-coded defects causing typical syndromes are found more often in adults, mutations of nuclear genes causing variable symptoms predominate in children (Table 17.3
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


