23
Metabolic Disorders Not to Miss
Introduction
Metabolic epilepsies are rare but important disorders that, in aggregate, are the subject of a significant proportion of child neurology. This chapter presents treatable metabolic epilepsies. Emphasis is given to entities in which prognoses are closely linked to early diagnosis and in which timely and targeted treatment is potentially crucial to avoid an otherwise catastrophic outcome.
Specific treatable metabolic epilepsies
Biopterin synthesis defects
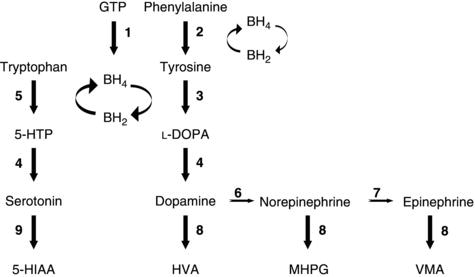
Disorders of synthesis or recycling of tetrahydrobiopterin (BH4), a vital cofactor in the synthesis of the monoamine neurotransmitters (e.g., dopamine, norepinephrine, epinephrine, and serotonin; Figure 23.1), can result in intellectual disability, epilepsy (typically myoclonic seizures with childhood onset), and extrapyramidal manifestations, including rigidity and dystonia. Notably, neuroimaging may show basal ganglia calcifications in this group of disorders, which may be reversible with folinic acid therapy. Biopterin disorders can usually be identified by hyperphenyalaninemia on newborn screening, although some variants are associated with a normal blood phenylalanine. Thus, evaluation for disorders in the BH4 pathway should be performed in infants with unexplained neurological disease. Treatment options include BH4 supplementation, 5-hydroxytryptophan as a precursor to serotonin, L-dopa, and monoamine oxidase or catechol-O-methyltransferase (COMT) inhibitors.
 CAUTION!
CAUTION!Cerebral folate deficiency
Cerebral folate deficiency is not a strictly defined syndrome but may represent a common final pathway of different neurological and genetic conditions. One underlying cause of cerebral folate deficiency involves autoantibodies that affect the folate FR1 receptor, which is required to transport folate from plasma to CSF. The phenotypes associated with cerebral folate deficiency involve epilepsy, intellectual disability, developmental regression, dyskinesias, and autism. Optic atrophy and cortical visual loss have been observed. Clinical diagnosis is based on low CSF 5-methyltetrahydrofolate, with normal peripheral folate levels. Treatment with folinic acid, which has better blood–brain barrier entry than folate, has been reported to improve seizures and other neurological dysfunction.
Figure 23.1. Monoamine and serotonin synthesis pathway. Reproduced with permission from Pearl, PL. Inherited Metabolic Epilepsies. New York: Demos Medical Publishing, 2013.
Biotinidase deficiency
Biotinidase deficiency can present with intermittent metabolic acidosis and an organic acid profile of lactic and propionic acidemia. The phenotype is developmental delay, hypotonia, seizures, ataxia, and rash. Signs and symptoms usually appear between 3 and 6 months after birth, when the prenatal supply of biotin is exhausted. If left untreated, the deficiency can lead to cerebral edema and permanent injury. Treatment with 10 mg/day of biotin has resulted in gratifying outcomes, although sensorineural hearing loss and optic atrophy with vision loss, once present, tend to persist. Early and sustained therapy can prevent the onset of symptoms and subsequent neurological deficits.
 SCIENCE REVISITED
SCIENCE REVISITED CAUTION!
CAUTION!Serine synthesis defects
Disorders of serine synthesis present with a phenotype of congenital microcephaly and psychomotor retardation, which may be nonspecific, likely leading practitioners to suspect in utero processes such as a TORCH infection (infection with toxoplasmosis, syphilis, rubella, cytamegalovirus or herpes simplex virus) or other causes of perinatal, static difficulties. Yet a treatable metabolic disorder would be important to consider. While rare, there are disorders of serine biosynthesis that may be amenable to supplemental serine and glycine, in which prenatal intervention can prevent manifestations in newborns. Diagnosis of serine synthesis disorders can be confirmed by amino acid analysis of plasma or CSF for low serine.
 SCIENCE REVISITED
SCIENCE REVISITEDCreatine synthesis and transport deficiencies
Metabolic disorders of creatine were first described in 1994 with the discovery of guanidinoacetate methyltransferase (GAMT) deficiency. The phenotype includes failure to thrive and early developmental delay, neurological regression, intellectual disability, autistic behavior, hypotonia, epilepsy, movement disorders, and abnormal pallidal signal on MRI. MR spectroscopy may suggest the diagnosis by detection of a low creatine peak. Creatine disorders resulting from GAMT or arginine:glycine amidinotransferase (AGAT) deficiency can be treated with creatine supplementation, along with arginine restriction and ornithine supplementation in the case of GAMT deficiency. Some individuals will need complementary antiepileptics, although others may respond to creatine supplementation alone.
 SCIENCE REVISITED
SCIENCE REVISITEDRelated posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


