and Aditya G. Shivane1
(1)
Cellular and Anatomical Pathology Level 4, Derriford Hospital, Plymouth, UK
Abstract
The functioning of nervous system can be affected by disturbances in metabolic substrates such as glucose and electrolytes, accumulation of toxic substances and deficiency of essential nutrients such as vitamins. This group of diseases are clinically important because correction of the metabolic disturbance may result in restoration of normal function. The majority of these conditions have relatively non-specific neuropathological features, but some conditions have characteristic gross and microscopic pathology. The clinically relevant conditions and those with characteristic neuropathology are discussed in detail.
Keywords
Metabolic diseasesEncephalopathyToxinsAlcoholVitaminsThe nervous system is biochemically very complex and quite sensitive to alterations in the internal milieu resulting from derangement in metabolites, exposure to toxins and deficiency of essential nutrients. The brain dysfunction secondary to these biochemical alterations, in most cases, results in non-specific neuropathological changes. However, there are some conditions associated with characteristic gross and microscopic changes within the nervous tissue. There may also be some regional variation or selective vulnerability of different brain regions in these diseases, partly explained by vascular patterns. This chapter discusses some of the common metabolic, toxic and nutritional diseases, some of which have characteristic neuropathology. This group of diseases are clinically important to diagnose in life because correction of the metabolic derangement may restore function.
11.1 Metabolic Diseases
11.1.1 Hypoglycaemia
The nervous tissue depends solely on a continuous supply of glucose for their energy. The stores of glucose and glycogen are very small and therefore, severe and prolonged periods of hypoglycaemia result in brain damage. The important causes for hypoglycaemia include- excess of exogenous insulin/or hypoglycaemic drugs, primary hyperinsulinism due to an islet cell tumour, liver disease, adrenal insufficiency and nesidioblastosis (in neonates). The common symptoms of hypoglycaemia include headache, confusion, irritability, incoordination, and lethargy, leading to stupor and coma. The MRI may show signal changes in temporal, occipital, insular cortex, hippocampus, basal ganglia, and deep white matter, usually sparing the thalami. In acute hypoglycaemia the brain may only show congestion and mild swelling. The microscopic changes include selective neuronal degeneration from the following brain regions- hippocampus (CA1-subiculum, CA3, CA4), cerebral cortex (layers 3, 5 and 6), caudate, putamen and dentate nucleus (in infants). Unlike hypoxia-ischaemia, the Purkinje neurons in cerebellum are spared. In chronic hypoglycaemic injury the brain may show cortical thinning, atrophy of hippocampi, white matter, caudate nucleus and putamen. The histology shows laminar neuronal loss and gliosis in the cortex. Overall, the brain changes in hypoglycaemia are similar, but not identical, to those seen in hypoxic-ischaemic injury [1, 2].
11.1.2 Electrolyte Imbalance
Amongst all the different serum electrolytes, the disturbances in the sodium and calcium are the most common and cause significant morbidity.
The causes for hyponatremia are varied and include- excess water ingestion/intravenous infusion of fluids, syndrome of inappropriate anti-diuretic hormone secretion (SIADH), diuretics, adrenal insufficiency, hypothyroidism, cirrhosis, renal or cardiac failure. The symptoms of hyponatremia include lethargy, headache, nausea and vomiting, leading into seizures, coma and death. The brain is swollen due to intracellular accumulation of fluid. Rapid correction of hyponatremia by infusion of hypertonic saline may result in a monophasic demyelinating condition affecting pons (central pontine myelinolysis) (Figs. 11.1 and 11.2a), and/or extra-pontine structures (extra-pontine myelinolysis, Fig. 11.2c) now termed ‘osmotic demyelination syndrome’ (ODS) [3].
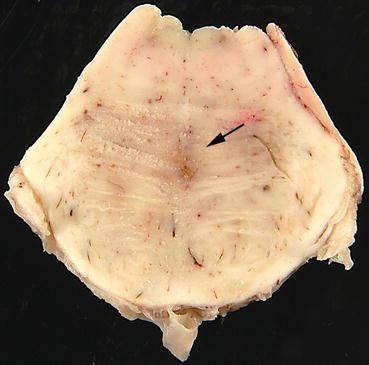
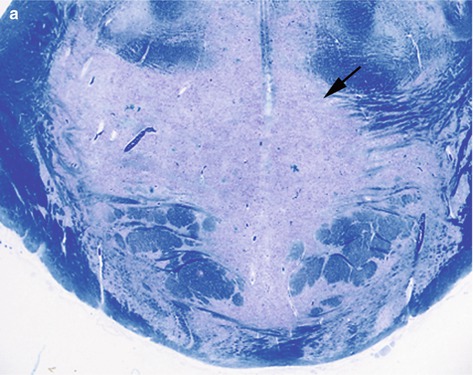
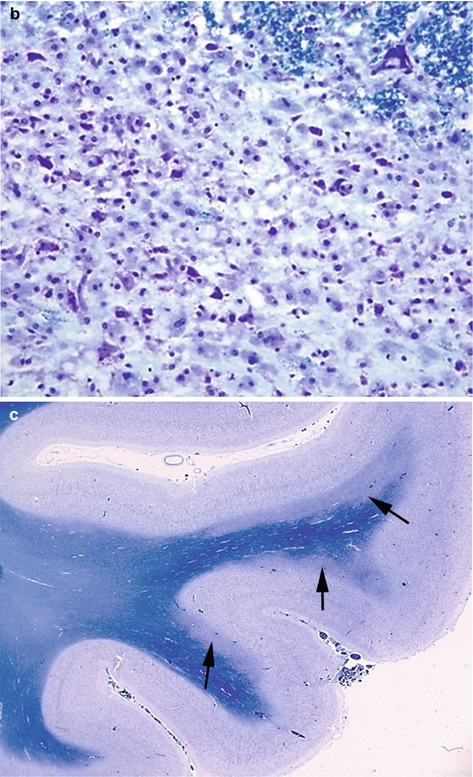
Fig. 11.1
Transverse section of mid pons from a case of central pontine myelinolysis showing an ill-defined central area of grey discolouration (arrow) within the basis pontis
Fig. 11.2
(a) Histology showing a large pale area of demyelination (arrow) within the basis pontis, (b) showing a dense infiltrate of foamy macrophages, (c) areas of demyelination in the temporal lobe (arrows) (extra pontine myelinolysis). LFB/CV stain
Conditions like alcoholic liver disease, burns, SIADH, hyperemesis gravidarum and psychogenic polydipsia predispose to ODS and the incidence is high in liver transplant patients. Many cases are fatal, but partial or complete recovery is possible. The pontine lesions appear granular, soft and gray discoloured to the naked eye, but are better delineated with stains for myelin. The common extra-pontine sites include cerebellum, lateral geniculate body, internal, external and extreme capsule, subcortical white matter, basal ganglia and thalamus. The histology confirms areas of demyelination with infiltration by lipid-laden foamy macrophages, but scanty lymphocytes (Fig. 11.2b). The axons are well preserved.Hypernatremia results from inadequate replacement or excessive loss of water especially in burns patients, diabetes insipidus, osmotic diuresis or excessive salt ingestion. Symptoms include confusion, lethargy, stupor, seizures or rarely coma. Neuropathology may show venous thrombosis and haemorrhage.
Disturbances in calcium metabolism include Fahr’s disease (familial idiopathic basal ganglia calcification), focal calcification in association with various infections, metabolic, endocrine and genetic disorders, and hypercalcemic encephalopathy. Fahr’s disease shows autosomal dominant or recessive inheritance with onset of symptoms around 30–60 years. The calcification is much more extensive than that seen in old age and involves basal ganglia, cerebral sulci, and dentate, subthalamic and red nucleus. The calcification of the blood vessels may lead to ischaemia. Hypercalcemic encephalopathy results from primary hyperparathyroidism and cancers with extensive bone involvement. The typical symptoms include confusion, seizures, headache and visual disturbance. The imaging shows symmetrical changes in the posterior cortical and subcortical regions, termed ‘posterior reversible encephalopathy syndrome’ (PRES) [4]. The pathology has revealed cerebral oedema in the white matter of parietal and occipital lobes [5].
11.1.3 Hepatic Encephalopathy
Hepatic encephalopathy is an acquired condition which results from severe liver disease or porto-caval shunting. Raised levels of ammonia in blood, seen in these conditions, cause astrocytic injury. The early symptoms include inattentiveness and short-term memory loss which progress to confusion, flapping tremor (asterixis), drowsiness, stupor and coma. Patients have characteristic breath odour (fetor hepaticus). The acute form can be rapidly fatal. The neuropsychiatric and motor symptoms associated with chronic or repeated episodes of encephalopathy disappear after liver transplantation. The neuropathology shows presence of abnormal astrocytes within deep cortical layers, basal ganglia, diencephalon, cerebellar dentate nucleus and brainstem. The abnormal astrocytes have enlarged, vesicular, round or lobulated nuclei with marginated chromatin and little or no cytoplasm. These are termed ‘Alzheimer type II astrocytes’ (see Chap. 2). Alzheimer type II astrocytes are not specific for hepatic encephalopathy and can be seen in various other acquired metabolic disorders. Chronic cases show neuronal loss, gliosis and microcavitation. Corticospinal tract degeneration may be noted within the spinal cord which results in hepatic myelopathy.
11.1.4 Wilson’s Disease
Wilson’s disease (also termed ‘hepatolenticular degeneration’) is an uncommon treatable disorder of copper metabolism with autosomal recessive pattern of inheritance. The disease is caused by mutation in a copper-transporting ATPase gene (ATP7B) encoded on chromosome 13, which is required for export of copper from the cell. The copper accumulates in the liver, brain and cornea leading to cirrhosis, neurological dysfunction and Kayser-Fleischer rings. The neurologic features include extrapyramidal movement disorder, spasticity, coarse tremor, dysarthria and dementia. Biochemical changes include low serum ceruloplasmin (copper-transporting protein), increased copper in the liver and decreased urinary excretion of copper. Macroscopic examination of brain shows shrinkage, cavitation and brown discolouration of putamen and caudate nucleus. These affected areas show neuronal loss, gliosis and pigment-laden macrophages. Alzheimer type II astrocytes and Opalski cells (small astrocytic cells with dark nuclei and intense pink cytoplasm) are common. The excess copper accumulation within the tissues is prevented by using chelating agents.
11.1.5 Uraemic Encephalopathy
Uraemic encephalopathy develops in patients with renal failure. It is believed to be caused by accumulation of toxic metabolites including urea and alterations in neurotransmitters. The symptoms range from mild cognitive changes to delirium and coma. Peripheral neuropathy is common. The neuropathology shows non-specific abnormalities which include cerebral atrophy, gliosis and foci of perivascular demyelination. Rare cases may show central pontine or extra-pontine myelinolysis. Patients with end-stage renal disease on dialysis can present with two distinct disorders- dialysis disequilibrium syndrome (due to water intoxication) and dialysis dementia (due to aluminium toxicity).
11.1.6 Amino Acid Disorders
These are quite rare, mostly autosomal recessive disorders of amino acid metabolism presenting in neonatal life with various nonspecific symptoms and signs. There are several biochemical screening tests which help in making an early diagnosis. This is important because prompt and early treatment with dietary manipulation can prevent future irreversible brain damage. Table 11.1 highlights the salient features of common amino acid disorders.
Table 11.1
Aminoacid disorders
Condition | Defect | Accumulated product | Neuropathology |
|---|---|---|---|
Phenylketonuria (PKU) | Phenylalanine hydroxylase (PAH) gene | Phenylalanine | Spongiosis of white matter, gliosis and delayed myelination |
Hyperglycinaemia | Glycine cleavage system (GCS) | Glycine | As above. Reduced white matter volume. Vacuolating myelinopathy |
Maple syrup urine disease | Mitochondrial branched-chain alpha-ketoacid (BCKA) dehydrogenase complex | BCKA and BCAA (branched-chain amino acid) Peculiar odour in urine | Spongiosis and gliosis of white matter, aberrant orientation of neurons, abnormal dendrites and dendritic spines Rarely acute axonal neuropathy |
Homocystinuria | Cystathionine beta-synthase deficiency (methionine pathway) | Homocystine | Thromboembolic disease with multiple cerebral infarcts |
11.1.7 Urea Cycle Disorders
This group of disorders cause hyperammonaemia as a result of failure to convert ammonia into urea. They can be divided into disorders of the enzymes of the urea cycle and disorders of the transporters or metabolites of amino acids related to the urea cycle. Ornithine transcarbamylase deficiency, an X-linked condition, is the most common disorder in this group. The neuropathological changes can vary from a normal appearing brain with Alzheimer type II astrocytic change to severe cortical and deep grey matter damage.
11.1.8 Porphyrias
Porphyrias are a group of disorders of haem biosynthetic pathway which result in overproduction and excretion of porphyrins. The overproduction may be within the liver or blood. Only the hepatic forms (acute intermittent porphyria and porphyria cutanea tarda) result in neurological disease which include peripheral autonomic and motor neuropathy, and psychiatric symptoms. Neuropathological changes in the brain are variable or minimal and may be related to associated hypoxia/ischaemia. The anterior horn cells of the spinal cord may show chromatolysis and there may be degeneration of spinal tracts. The peripheral nerves may show axonal neuropathy and demyelination.
Related posts:






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


