Fig. 5.1
CE usually develops within a short time period of hours to days and involves strong neuroinflammatory alterations within CSF spaces and the brain parenchyma. The latter has been demonstrated in neuroimaging in a subgroup of cases only (compare Osborn et al. 2010)
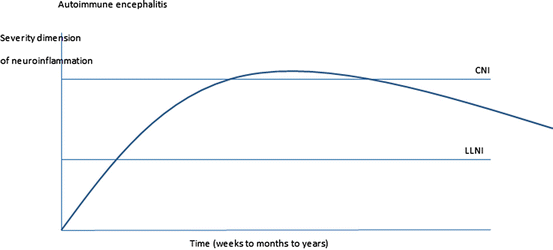
Fig. 5.2
AE includes prodromal symptoms over weeks, followed by more or less severe neurological symptoms after some weeks and variable, not infrequently long, recovery times (compare Dalmau et al. 2011; Najjar et al. 2013; Prüss 2013; Peery et al. 2012; Lancaster et al. 2011). In principle the graph resembles CE in Fig. 5.1, but the timeline, especially of the recovery phase, is more extended, and acute neuroinflammation is less severe
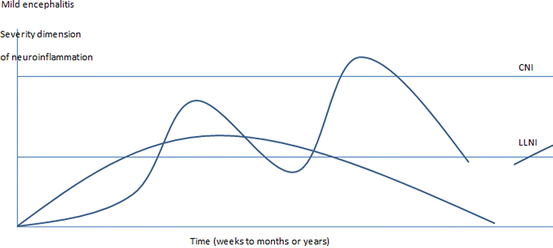
Fig. 5.3
The ME model is based on experimental findings that demonstrated slow onset and generally minor or localized classical encephalitis, the latter only poorly reflected in clinical symptoms. Human ME cases will often show normal brain images and normal CSF findings, if advanced methods are not used. Minor LLNI stages might persist for a long time before symptoms develop and coincide with long prodromal stages, as observed in schizophrenia (Häfner 1995; A. Riecher-Rössler). A similarity between basic symptoms in schizophrenia as described by Gerd Huber and initial symptoms in LE is worth noting (review in Bechter 2002). The major aspect of the ME definition is that some LLNI state should be detectable in a cross-sectional assessment during the active disease phase, especially by CSF examination. ME may not simply reflect secondary effects from systemic inflammation without any autochthonous neuroinflammation. Contributing factors may be many, not least neurodevelopmental alterations (Müller and Bechter 2013)
Some Historical Remarks on Etiology Research in Psychiatric Disorders
After about 50 years of strong rejection and some additional 20 years of debate, the initial hypothesis that syphilis might represent an important risk factor for general paresis was accepted, and later turned into a strict view. The spirochete, although detected in the brain of only very few cases, represents the infectious cause of general paresis (GP) (compare Bechter 1995). A major factor contributing to the acceptance of the causality relationship in general was the similarity of findings in late syphilis with findings in the brains of dogs with experimental trypanosomiasis, as well as the successful treatment of the disease with malaria (and later penicillin). Also very important was considerable progress at the time in laboratory diagnosis with the introduction of the Wassermann reaction (a preliminary type of antibody testing) and of lumbar puncture and CSF analysis. To imagine the relevance of this breakthrough, one should recognize that, at the time, GP was very frequent and often fatal. For example, in Hamburg around 1900 about 20 % of psychiatric inpatients suffered from GP. Thus, the new principle of understanding GP as a late stage of an infectious disease was like a revolution. This also triggered the idea of searching for other infectious agents causing severe psychiatric disorders like schizophrenia, but this turned out to be difficult. Today, many evidently think that the pathogenesis of GP is very clear and causal. This is apparent in remarks from many colleagues who, when they are initially presented with the ME hypothesis, nevertheless view it critically or reject it. Yet, when we go into great detail about the present knowledge of the pathogenesis of GP, it appears that, in a strict scientific sense, our understanding of the pathogenesis remains quite limited. For example, we cannot explain why, after a long latency of years, GP develops only in a limited subgroup of the infected. This is a core problem of medical understanding that is involved in the further discussion of the ME theory here.
A similar problem was gastroduodenal ulcer disease in people infected with Helicobacter pylori (see Marshall 2006). Indeed, many infections are characterized by low overall pathogenicity (number of diseased per number of infected). Such was also the case with poliomyelitis, raising the debates over 20 years: only 2 % of patients get the terrible paresis but many show the serum antibodies. The same is true for EBV infection, CMV infections, or borreliosis (Bechter 2013). The recent studies on risk factors in schizophrenic and affective spectrum disorder represented by infections and autoimmune disorders and brain trauma (Benros et al. 2012, 2013, 2014; Sörensen et al. 2014) are therefore extremely important, because conclusive epidemiologic data from the Danish National Register provide a basis for reinforced clinical studies. These specific risk factors nicely match with the ME theory. But one should be aware of considerable problems with such research, when treating a patient. This is again highlighted by looking back into the history of GP: At the end of the twentieth century, leading newspapers unjustifiably criticized the awarding of the Nobel Prize to Julius Wagner-Jauregg for the introduction of malaria therapy. This appears to have been related to the often observed tendency to simplify questions of causality. The story of malaria therapy, then fever therapy, and later penicillin, raised a hype to search for other infectious causes of psychiatric disorders. The search remained unsuccessful for a long time. A similar revival followed after the description of the first slow virus infection by Gajdusek and others (Gajdusek 1965; Kurstak 1991; Bechter 1995).
Multiple Interacting Systems and Factors
Severe psychiatric disorders of the affective and schizophrenic (including bipolar) spectrum are complex diseases where many factors should be considered: certainly genes, age of onset, timing, stress, infections and autoimmunity, and possibly chance (Drexhage et al. 2010; Gibney and Drexhage 2013; Hertz et al. 2013; Rector et al. 2014). The situation is rather similar to systemic autoimmune disorders, where the idea is prevalent that “every autoimmune disorder has to be considered as of infectious origin unless otherwise proven” (repeated public remarks by Yehuda Shoenfeld, congress president at the ninth International Autoimmunity Congress, Nice, France, March 26–30, 2014), outlined in more detail in textbook “Infection and Autoimmunity,” (Shoenfeld and Rose 2004).
Interestingly, in many systemic autoimmune disorders, there is a considerable subgroup of patients suffering from comorbid psychiatric symptoms. In some, there are even very severe psychoses over certain time periods, for example in lupus erythematosis (Diamond and Volpe 2012) and especially in cases with antiphospholipid autoantibodies (D’Ippolito et al. 2014). Time and duration of pathology is also definitely involved, as evidenced, for example, in multiple sclerosis (Rossi et al. 2014). Relevance of time was also directly investigated: the duration of systemic inflammation elicited different effects over the long term as compared to the short term (Maggio et al. 2013). But pathomechanisms are difficult to assess in individual cases. Risk genes of psychiatric disorders may be directly implicated in the life cycles of specific pathogens (Carter 2009). Some infections may be relevant in people with a specific genetic background (Kumarasinghe et al. 2014), or a specific genetic make-up like sphingomyelinase-ceramide system in depression (Gulbins et al. 2013), known to be involved in inflammatory responses. At-risk states showing an inflammatory molecular signature in both CSF and blood are associated with infectious agents in schizophrenia; however, abnormalities were even exacerbated in risk subjects (Hayes et al. 2014). Both systemic and CNS-specific tryptophan pathways appear to be involved. This has been demonstrated not only for IDO and cytokines, but also protective factors and cannabinoid receptor; all of these findings are linked to an activated inflammatory response (Myint and Kim 2014; Brietzke et al. 2009; Anderson et al. 2013; Sánchez-Blázquez et al. 2014). Stress represents another player, for example in psychosocial factors (Meyer-Lindenberg 2010). A relatively well-defined neurological autoimmune disorder, Guillain–Barre syndrome, associated with preceding Campylobacter jejuni infection, is frequently accompanied by psychiatric symptoms that include psychosis (Schielke et al. 2014). New insights into the role of the gut microbiome and the immune system of the host suggest a continuous battle taking place, with possible strong influences on CNS disorders (Wang and Kasper 2014). This also holds true for experimental autoimmune encephalomyelitis (Berer et al. 2011, 2014).
The role of prenatal infections has been reviewed repeatedly and the increase in risk from various specific infectious agents was considerable (Brown 2011), though not specific for schizophrenia but rather a spectrum of psychiatric syndromes (Bechter 2013). A set of experimental findings on infections during the intrauterine life showed subsequent disturbances of neurodevelopment or of immune development with schizophrenia-like symptoms in adulthood (Meyer 2013; Juckel et al. 2011). A recent model demonstrated ongoing inflammation within the brain, supporting the ME theory (Mattei et al. 2014). It should be recognized that neither models nor humans studies show an association between neurodevelopmental abnormalities and specific disorders. For example, minor neurodevelopmental abnormalities are also found in depression (Whittle et al. 2014). This points to the lack of specificity of brain seizures and strokes, with the consequence of overlapping scenarios (see also Buchsbaum and Rieder 1979; Bechter 2012). The recently discovered inflammasome is also involved in neuroinflammation, but is only beginning to be studied (Walsh et al. 2014). This will surely have relevant influences on microglia, which represents an important CNS cell system, relevant not only in pathology but also in normal physiology and in the developing brain (Kettenmann et al. 2013). Activation of microglia has been demonstrated in these psychiatric disorders (discussed in another chapter of this book).
Overall, there are many pathways and risk factors involved in the pathophysiology of LLNI, the basis of the psychiatric disorders in question here. Apparently, the scenario is extremely complex and difficult to dissect. To understand LLNI as either one common final pathway or alternatively several LLNI types or states appears plausible. Though it remains to be better defined, LLNI has the power to elicit and explain various symptoms or psychiatric disorders observed. The ME theory suggests that a subgroup of severe psychiatric disorders showing signs of LLNI are causally related to LLNI, although associated with varying symptom patterns, depending upon contributing and preexisting factors, e.g., genetic factors and many others (Bechter 2013; Müller and Bechter 2013).
Brain Barriers and Systemic Crosstalk
Systemic inflammation and inflammatory signaling can directly influence brain functions (Thomson et al. 2014) and elicit a pattern of symptoms described as sickness behavior (Dantzer 2012), in part fitting with symptoms observed in psychiatric spectrum disorders discussed here as the main candidates of ME theory. But sickness behavior cannot explain many aspects of these diseases nor their courses. Apparently, complex pathogenetic events are related plausibly to neuroinflammation. Of special interest here are recent insights from experimental neuroimmunology on the pathogenesis of chronic neuroinflammation (Schwartz and Baruch 2014): in general, acute inflammation needs to be actively resolved; otherwise, it becomes chronic, and this seems to be true also for neuroinflammation. The choroid plexus (CP) represents an interface between blood and CSF spaces; immune cells are recruited and educated at the CP and then released into the CSF. But there are several brain barriers. These barriers must be explained briefly here. One should differentiate between the blood–brain barrier (BBB) and the blood–CSF barrier (BCSFB). The BCSFB is defined as an anatomical structure, the CP, or the CP plus circumventricular organ (Schwartz and Baruch 2014; Wolburg and Paulus 2010).
The BBB is generally regarded as representing a barrier between brain vasculature and brain parenchyma (Engelhardt and Ransohoff 2012). However, the function of the BBB has to be further differentiated, one for cells and one for solutes, with anatomically differing sites (Bechmann et al. 2007). In clinical terminology, blood–CSF barrier (BCB) dysfunction is used differently. One paper (Reiber and Peter 2001) describes certain abnormal CSF findings, in effect a proportional albumin increase, which represents an increase of blood-derived proteins within the CSF. BCB dysfunction may mainly relate to blood CSF flow pattern changes or CSF production (Reiber 1994; Seyfert and Faulstich 2003; Seyfert et al. 2004, 2009 ), but not true dysfunction of the anatomically defined BCB or BBB. In addition, in such a complex scenario a new pathway has to be included, the so-called glymphatic pathway of CSF through the brain, characterized by periarterial inflow and perivenous outflow of CSF (Benveniste et al. 2014; Iliff et al. 2012). The CSF flow pulsates dramatically from the ventricles to the subarachnoid spaces and back; the flow distance is about 10 cm per pulse back and forth at the aqueduct (Gupta et al. 2009, 2010; Kurtcuoglu 2012). It has so far been only partially investigated, and poorly down the neuraxis. In addition, the pulsating brain drives the flow of the extracellular CNS fluid, the flow has been studied in some detail and described as volume transmission mode balancing the wiring transmission (the latter mainly represented by synaptic transmission). This exchange includes not only molecular but also particulate fluid contents, e.g., exosomes (Fuxe et al. 2013). Understanding the CSF flow patterns and their respective influences on CSF content concentrations is of prime importance.
The long known absorption pathway of CSF to arachnoid granulation is not the only one. There is considerable outflow through the area called cribrosa to nasal submucosa and then to the cervical lymph nodes (Carare et al. 2014) and along all brain nerves and peripheral nerves into peripheral tissues (Bechter 2011). Interestingly, CSF cells can also follow these CSF outflow pathways through the area, cribrosa (Kaminski et al. 2012), and along lumbar nerves (Schmitt et al. 2011). Signal transmission by extracellular vesicles is now known to be relevant for CSF immune response (Robbins and Morelli 2014). The role of CSF cells as an independent player has been newly introduced. Immune cells apparently support normal brain functions, e.g., they support the cortex and cognitive functions directly (Kipnis et al. 2012; Baruch et al. 2013). CSF cells educated at the CP play a critical role in the interaction between neuroinflammation and neurodegeneration (Schwartz and Baruch 2014). This latter point is very interesting with regard to the historical controversies about the pathology of general paresis (see also above): the histopathologists in particular rejected the hypothesis that syphilis was a possible risk factor for general paresis (GP), not least because they preferred to distinguish between inflammatory and neurodegenerative GP (Review in Bechter 1995).
Outlook
From first ideas formulated as the mild encephalitis hypothesis, some experimental treatment approaches based on these ideas were developed. CSF filtration was adopted from neurological research, where it had been successful in treating therapy-resistant Guillain–Barre syndrome. Its use resulted in a considerable improvement over the long term in cases of therapy-resistant schizophrenia or depression, though only in a small number of cases (Bechter et al. 1999, 2000; Bechter 2007). Skepticism of many colleagues at that time about such experimental treatment approaches was evident. Now one can notice a dramatic change of scientific mainstream thinking. Many researchers accept the possibility that a considerable number of subgroups of severe psychiatric disorders include some neuroinflammatory process, which may at least contribute to, if not causally underlie, these disorders. However, the pathogenesis remains incompletely understood. Previous theories were based on specific or single neurotransmitter or receptor models. The ME theory also takes a simplified point of view by defining as a main criterion the ability to detect some LLNI process at a relevant stage of the disease in the individual patient. Such an approach seems nevertheless to be required in the clinic. Risk factors or contributing factors are an additional prerequisite. The ME theory fits with such aspects as increased risk through intrauterine injuries, through neurodevelopmental abnormalities or through immune developmental abnormalities, especially with the risk increase by infectious and autoimmune disturbances and head trauma during lifetime. These latter findings are in accordance with the association between infections and autoimmune disorders in general. The investigation of specific triggers of ME in an epidemiological scenario with respect to psychiatric disorders represents important progress. However, it remains a challenge to investigate the role of specific infections in the individual case, and to differentiate LLNI processes, perhaps by neuroimaging, CSF investigation and other new methods (Bechter 2013). Methods need to be improved. Newly combined methods like the TMS/EEG may improve direct insight into the specific pathology associated with certain symptoms. With combined long-term studies it may be possible to differentiate between different patterns of brain deficits, such as between normal age effects and prodromal and illness-related alterations (Kumarasinghe et al. 2014). Molecular profiling in blood and CSF may be helpful (Herberth et al. 2014). New pathways like microRNA dysregulation may need to be investigated.
In summary, psychiatric research should focus on etiopathogenetic research on LLNI and the ME theory.
References
Anderson G, Berk M, Dodd S, Bechter K, Altamura AC, Dell’osso B, et al. Immuno-inflammatory oxidative and nitrosative stress and neuroprogressive pathways in the etiology, course and treatment of schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2013;42:1–4.PubMed
Baruch K, Ron-Harel N, Gal H, Deczkowska A, Shifrut E, Ndifon W, et al. CNs-specific immunity at the choroid plexus shifts toward destructive Th2 inflammation in brain aging. Proc Natl Acad Sci U S A. 2013;110(6):2264–9.PubMedCentralPubMed
Bechmann I, Galea I, Perry VH. What is the blood–brain barrier (not)? Trends Immunol. 2007;28(1):5–11.PubMed
Bechter K. Research strategies in “slow” infections in psychiatry. Hist Psychiatry. 1995;6:503–11.PubMed
Related posts:
 Experimental Human Endotoxemia, Sickness Behavior, and Neuropsychiatric Diseases
Developmental Immune Activation Models with Relevance to Schizophrenia
Role of Autoimmunity and Infections in Tourette Syndrome
The Role of Inflammation in Alzheimer’s Disease
Experimental Human Endotoxemia, Sickness Behavior, and Neuropsychiatric Diseases
Developmental Immune Activation Models with Relevance to Schizophrenia
Role of Autoimmunity and Infections in Tourette Syndrome
The Role of Inflammation in Alzheimer’s Disease
 The Role of Infections and Autoimmune Diseases for Schizophrenia and Depression: Findings from Large-Scale Epidemiological Studies
The Role of Infections and Autoimmune Diseases for Schizophrenia and Depression: Findings from Large-Scale Epidemiological Studies
 Animal Models Based on Immune Challenge: The Link to Brain Changes and Schizophrenia
Animal Models Based on Immune Challenge: The Link to Brain Changes and Schizophrenia
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


