Disease/syndrome
Clinical features
Genetics
Prognosis
Defects of mtDNA
Chronic progressive external ophthalmoplegia syndrome (CPEO)
Mimicking KSS plus
AD; AR
Progressive disorder of variable severity, and phenotype. Onset: late teens-early 20s
Kearns–Sayre syndrome (KSS)
Slowly progressive multisystem disease, onset <20 years
Mitochondrial
Death: most often in the third-fourth decade (see text)
Leber’s hereditary optic neuropathy (LHON)
Mean onset: 20 years with subacute loss of vision
Most common mtDNA mutations: 11778, 3460, 14484
Almost complete blindness in about 12 months
Mitochondrial encephalomyopathy lactic acidosis and stroke-like episodes (MELAS)
Progressive neurodegenerative disorder. Onset: 2–15 years
mtDNA point mutations: A3243G (80 % of the cases)
Death: 10–35 years of age (see text)
Myoclonic epilepsy and ragged-red fiber disease (MERRF)
Myoclonus, epilepsy, ataxia, muscle weakness, deafness, and dementia. Onset: childhood or early adulthood
Mostly caused by the 8344A > G mtDNA mutation (over 80 % of the cases)
Variable, but progressive phenotype. Death: usually between 30 and 40 years of age (see text)
Neuropathy, ataxia, and retinitis pigmentosa (NARP)
Onset: childhood or early adulthood
NARP syndrome is maternally inherited. Mutated mtDNA is >95 %, patients show the maternally inherited LS
Severity depends on the mutation load. Patients may become blind, deaf, demented, and wheelchair bound
Subacute necrotizing encephalomyelopathy (Leigh syndrome/LS)
Psychomotor delay, seizures, dyspnea, ophthalmoparesis, nystagmus, vomiting. Onset: infancy or childhood (can also occur in teens and adult patients)
May be: sporadic, due to mtDNA mutations (10–30 %)
Poor prognosis: individuals typically live from a few years to mid-teens
Defects of nuclear DNA
Myoneurogastrointestinal disorder and encephalopathy (MNGIE)
Ptosis, PEO, gastrointestinal dysmotility, neuropathy, leukoencephalopathy. Onset: late teens
AR
Most patients die before age 40 (see text)
Coenzyme Q10 deficiency disorders
Encephalomyopathy, myopathy, infantile multisystemic form, ataxic nephropatic disease, ataxic form.
AR
Variable prognosis associated with therapeutic response to coenzyme Q10 supplementation
Polymerase gamma mutations (POLG1 and POLG2)
AD and AR progressive external ophtalmoplegia; Alpers-Huttenlocher syndrome with epilepsy and hepatic failure; childhood myo-cerebro-hepatopathy; myoclonic epilepsy myopathy sensory ataxia (MEMSA); ataxia neuropathy spectrum (ANS) disorders
AD and AR
Poor prognosis for most POLG1 mutations with early clinical presentation (infancy and childhood). Some forms of adPEO, however, cause serious and critical neurological problems in the middle-adult age
Subacute necrotizing encephalomyelopathy (Leigh syndrome/LS)
Psychomotor delay, seizures, dyspnea, ophthalmoparesis, nystagmus, vomiting. Onset: infancy or childhood (can also occur in teens and adult patients)
May be: sporadic, or due to AD-AR, and X-linked nuclear gene mutations
Poor prognosis: individuals typically live from a few years to mid-teens
In terms of function, the RC carries out two main integrated reactions:
1.
The exoergonic transfer of electron equivalents from the reduced electron carriers NADH and FADH2 to molecular oxygen (respiration, complex I-IV), a process coupled to proton translocation across the inner membrane.
2.
The endoergonic ATP synthesis (phosphorylation, complex V) driven by the energy primarily stored as an electrochemical proton gradient [2, 3]. Electron transfer and ATP synthesis are coupled, or linked [3, 5], since the RC works as a proton pump which generates a proton gradient and a membrane potential of about 180 mvolts across the inner membrane and the proton gradient is utilized by the ATP synthase to phosphorylate matrix ADP. During this process the proton gradient is decreased and this activates respiration, i.e., electron transfer [3, 5].
OXPHOS diseases are mostly expressed in high-energy demanding nonmitotic tissues such as heart, skeletal muscle, and the central and peripheral nervous system [6].
The “competence” of the RC, i.e., the effectiveness of ATP generation compared to caloric intake, is called coupling efficiency and reflects both the efficiency by which complexes I, III, and IV convert the oxidation of reducing equivalents into Delta-P (membrane potential) and the efficiency by which complex V converts Delta-P into ATP. Tightly coupled OXPHOS indicates a coupling efficiency that maximizes ATP generation per calorie intake [3, 5]. Remarkably, all the proton-translocating complexes of OXPHOS (complexes I, III, IV, and V) must be balanced to ensure that one complex is not disproportionally permeable to protons and thus shorts Delta-P. This is achieved by having the core electron and proton transport genes retained on a single piece of non–recombining DNA, the exclusively maternally inherited mtDNA [3, 4].
Key Facts
Terminology and definitions – Mitochondrial disorders are clinical phenotypes associated with abnormalities of OXPHOS.
Clinical features – Vary from CNS involvement (encephalopathies, stroke-like episodes, seizures, ataxia, and dementia) to PNS and striated muscles deficits.
Diagnostic markers
–Blood – Mildly elevated CK (muscle involvement); raised L and L/P values (disorders of OXPHOS)
–CSF – Mildly elevated proteins and raised L and L/P values (CNS involvement)
–Genetics – Maternal (mtDNA primary mutations); AD, AR, X-linked (primary nDNA mutations)
–Imaging often not specific; stroke-like lesions (MELAS); signal abnormalities in the basal nuclei, and brainstem (LS)
–Muscle biopsy – RRF (MERRF, MELAS, NARP, MNGIE); Cox-deficient fibers (MNGIE)
Top Differential Diagnoses – Myastenia gravis (PEO), multiple sclerosis (LHON), metabolic encephalopathies (MERRF), metabolic and hereditary myopathies and cardiopathies
Prognosis
Principles of treatment – Therapy is usually only supportive.
Disability – Most MD are chronic and progressive often causing premature death.
20.2 Terminology and Definitions
Mitochondrial disorders (MD) (alias mitochondrial encephalomyopathies, mitochondrial myopathies, oxidative phosphorylation disorders) are clinical syndromes associated with abnormalities of the common final pathway of mitochondrial energy metabolism, OXPHOS [1]. They can be caused by mutations in mitochondrial DNA (mtDNA) or in nuclear genes (nDNA).
20.3 Epidemiology
MD affect at least 1/8000 of the general population [7], but about one in 200 healthy humans harbors a pathogenic mtDNA mutation that potentially causes disease in the offspring of female carriers [8]. The risk of recurrence of MD in the offspring of an affected mother with a single mtDNA deletion is 4.11 % [9].
20.4 Clinical Features
Mitochondrial diseases most often involve the central nervous system (CNS), peripheral nervous system (PNS), and striated muscles. Affected patients, may exhibit a mixture of multiple organ dysfunctions, or deficits limited to visual impairment, e.g., Leber’s hereditary optic neuropathy (LHON).
Some peculiarities of mitochondrial functioning play a crucial role in determining the clinical characteristic of mitochondrial disorders.
20.4.1 Phenotypes
Defective OXPHOS principally affects tissues with high-energy demand (central nervous system; skeletal and cardiac muscle; peripheral nerves; eye; and endocrine organs), frequently resulting in multisystem disorders [10] mainly involving post-mitotic cells and tissues. Hence, on practical grounds, a diagnostic procedure to ascertain a mitochondrial disorder should be settled as soon as we recognize in a patient a pathology affecting two or more unrelated tissues or organs with high respiratory rates [6].
20.4.2 Maternal Inheritance
Mitochondrial diseases caused by mutation of mtDNA are transmitted in a pattern of maternal inheritance (children of both sexes inherit only the mother’s mitochondrial DNA). Nuclear gene defects can also affect mtDNA, leading to secondary mtDNA mutations that can be transmitted as autosomal dominant (AD), autosomal recessive (AR), and X-linked traits and can mimic the features of disorders caused by primary mtDNA mutations.
Mutations of mtDNA mainly cause mitochondrial diseases in adults; nuclear gene (nDNA) alterations often produce the more severe infantile phenotypes.
20.4.3 Homoplasmy/Heteroplasmy and “Phenotypic Threshold Effect”
Homoplasmy means that all copies of the mtDNA are identical. This is the common condition of normal organisms. Heteroplasmy is the presence in the cell of a mixture of normal and mutant mtDNAs. The proportion of mutant to wild-type mtDNA (amount of heteroplasmy) is a central factor in the appearance and severity of mitochondrial diseases because phenotypic expression of a genetic mtDNA defect occurs only when mutant mtDNA exceeds a threshold level which fluctuates in tissues, and in single affected individuals, partly explaining the clinical unpredictability of mitochondrial disorders. Besides, human tissues incorporate somatic mtDNA mutations with age, possibly sustaining age-associated mitochondrial dysfunction.
At present, the concept of heteroplasmy is still valid in pathology, though we recognize now that mitochondria are dynamic in nature [11]. As a matter of fact, any given mitochondrion is not a discrete organelle because it will fuse or merge, soon or after in its life, with other mitochondria. Mitochondrial fusion not only results in the mixing of outer and inner membrane but also of the matrix content, including the mitochondrial DNA organized in the matrix in discrete units called nucleoids [3]. Fusion and the opposite event of mitochondrial splitting, dubbed fission, do not simply occur at cell division but are constant features of the cell life, so that the identity of any mitochondrion is transient. The relevance of this concept in mitochondrial genetics is patent as one of its major consequence is that a given mitochondrion within a heteroplasmic cell, i.e., a cell containing more than unique species of mitochondrial DNA, will likely contain both mutant and wild-type DNA [3, 11].
20.5 Diagnosis
20.5.1 Diagnostic Markers
Blood
– Routine analysis: neutropenia, thrombocytopenia, and anemia characterize some OXPHOS disorders. Diabetes mellitus is a common feature. Creatine kinase (CK) is mildly elevated with muscle involvement. Raised lactate (L), with L/pyruvate (P) = 10–20, points to altered pyruvate metabolism; raised lactate with L/P > 20 points to disorders of oxidative phosphorylation. The amount of amino and organic acids (blood and urine), acyl-carnitines (free and total in blood and urine), coenzyme Q level (blood) may be an important clue to diagnosis.
Heart
– Cardiac arrhythmias and hypertrophic cardiomyopathy should routinely be searched for.
Ears and eyes
– Sensorineural hearing loss; cataracts, retinitis pigmentosa, optic neuropathy, and visual field defects are frequent features of MD.
CSF
– Cerebrospinal fluid protein are increased in encephalopathies; L and P, L/P (see above).
Muscle biopsy
– Routine light microscopy, immunohistochemistry. Ragged red fibers (RRFs) may be found in MERRF, MELAS, and NARP. Cytochrome oxidase deficiency may be found in LS, etc.
MRI
– Can be nonspecific or reveal signal abnormalities in the lentiform and caudate nuclei, thalamus, brainstem in LS (Fig. 20.1); transient stroke-like lesions not confined to vascular territories in MELAS. T2/FLAIR: hyperintense bilateral lesions in subcortical white matter, thalamus, basal ganglia, and brainstem in KSS. Prominent leukoencephalopathy, usually sparing corpus callosum in MNGIE.
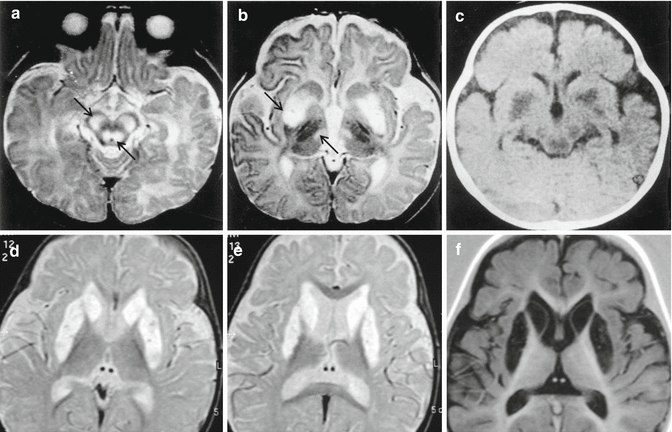
Fig. 20.1




Leigh syndrome. Two different cases. Top, a baby of 5 months; bottom, a child of 3 years. Top, axial T2-weighted images, (a, b), and axial CT, (c), demonstrate marked hyperintensities and hypodensity in cerebral peduncles, tegmen pons, bilateral putamen, and medial portion of the thalami (arrows). Bottom, axial T2-weighted images, (d, e), and axial IR image, (f), show necrosis of bilateral putamen and of the head of the caudate, visible as CSF cavitations of the nuclei
Related posts:


Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
