Molecular Basis of Select Childhood Psychiatric Disorders
Paul J. Lombroso
James F. Leckman
The past decade has seen remarkable progress in the application of molecular genetic strategies to the study of neurologic and neuropsychiatric disorders (1,2). The chromosomal location for a number of autosomal-dominant, autosomal-recessive, and X-linked disorders has been accomplished (Table 2.3.3.1). The genes that are mutated in many of these illnesses have been mapped or identified. Determining the function of both the normal and mutated proteins has begun. This chapter reviews some of the important accomplishments in this area. It begins with a discussion of the molecular bases for normal learning and memory and how mutations of key signaling proteins lead to developmental disorders, and concludes with a brief consideration of the molecular basis of additional child psychiatric disorders.
Learning and Memory
The ability to learn something new and then consolidate that information into long-term memories is part of normal development. As clinicians, we are often asked to evaluate whether a child is developing appropriately. Are specific skills emerging at the appropriate time points or are they delayed? Landmarks include the ability of a child to speak, to read, or to interact appropriately with peers. The mechanisms by which children learn to sit and crawl, walk and talk, and develop social skills have been the intensive focus of psychologists and psychiatrists over the years.
It is only in the past few decades that investigators have begun to study these questions at a molecular level (3,4). What has emerged is a fascinating story of how cells within the central nervous system communicate with each other during learning and initiate a series of events that lead to the formation of long-term memories. Equally interesting is the finding that disruptions to these normal processes contribute to several developmental disorders. We will first review some of the mechanisms by which normal learning develops before discussing disorders that occur when these processes are disrupted.
A central concept to emerge from research on learning and memory is that the formation of long-term memories requires both biochemical and structural modifications within neurons. The transformation of short-term, labile memories into more stable, long-term memories requires growth at specialized points of neuronal contact termed synapses. Synapses change shape as learning proceeds— new synapses are formed or old ones are strengthened. This phenomenon, termed synaptic plasticity, is found in all parts of the brain where memories are formed.
When an action potential arrives at the end of the axon, synaptic vesicles fuse with the presynaptic terminal membrane and release one or more of a variety of neurotransmitters. The transmitters diffuse across the narrow synaptic cleft and bind to receptors on the postsynaptic terminal. Neurons communicate in this way, and the subsequent processing of the incoming signal at the postsynaptic terminal results in long-lasting changes in both the shape and biochemical composition of synapses.
A tremendous amount of research has been devoted to understanding this process over the last decade (5). To summarize, a signal arrives at the surface of a neuron and induces the production of proteins required for the morphological and biochemical changes at the synapse. This experience-induced synaptic plasticity underlies how memories are formed, and the molecular mechanisms by which this occurs will be reviewed next.
A key player in these events is a member of the mitogen-activated protein kinase (MAPK) family of proteins, the extracellular-signal regulated kinase (ERK) (6,7,8). The arrival of a signal at the postsynaptic terminal activates a cascade of signaling proteins that leads to the activation of ERK (Figure 2.3.3.1). The ERK pathway is activated in all brain regions where synaptic plasticity occurs and is necessary for the formation of new long-term memories (5). This has been demonstrated by blocking the activity of ERK by injecting inhibitors of this enzyme into specific brain regions, such as the amygdala. The formation of memories that are normally consolidated within that structure does not occur (e.g., fear conditioning) (9,10). Similarly, if ERK activity is blocked within the hippocampus, the formation of memories that require the hippocampus is prevented (visual-spatial learning) (11).
ERKs are members of a family of enzymes termed kinases that add phosphate groups to substrate proteins. The addition of bulky, negatively charged phosphate groups often results in a change in the three-dimensional shape of the phosphorylated protein. The conformational change in turn often exposes a previously hidden amino acid domain buried deep within the structure of the protein. The exposure of the domain now permits interactions of the protein with other polypeptides or molecules further downstream in a cascade. For example, phosphorylation and activation of ERK leads in turn to the phosphorylation of a set of transcription factors. The transcription factors are now capable of initiating gene transcription (12).
In this way, a signal originating at the surface of a postsynaptic neuron is communicated to the interior of the cell, and a group of genes are transcribed. The proteins produced are necessary for the modifications at the spine that accompany synaptic plasticity. As a consequence, the neuron becomes more sensitive to future synaptic input: It responds with higher excitatory potentials than before. Moreover, the increase in responsiveness may last for prolonged periods of time, months or longer. This phenomenon, termed long-term potentiation, is thought to underlie the molecular changes that occur during the formation of stable, long-term memories (5,13).
A key point in the present discussion is that a series of proteins are “upstream” of ERK and necessary for its
activation, while a series of proteins are “downstream” of activated ERK and are required for transcriptional activation. It follows that disruption of components of this multistep pathway might in turn disrupt the development of synaptic plasticity. Mutations have recently been discovered in several genes involved in this pathway and these mutations lead to the three developmental disorders that are discussed next (Figure 2.3.3.1).
activation, while a series of proteins are “downstream” of activated ERK and are required for transcriptional activation. It follows that disruption of components of this multistep pathway might in turn disrupt the development of synaptic plasticity. Mutations have recently been discovered in several genes involved in this pathway and these mutations lead to the three developmental disorders that are discussed next (Figure 2.3.3.1).
TABLE 2.3.3.1 CHROMOSOMAL LOCALIZATION AND GENE ABNORMALITIES IN SELECTED PSYCHIATRIC AND NEUROLOGIC DISORDERS | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
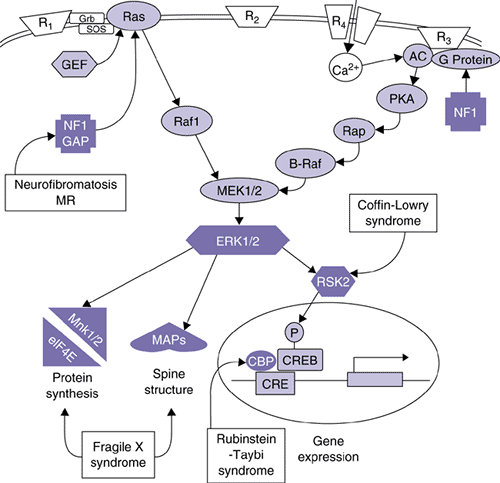 FIGURE 2.3.3.1. The ERK MAPK cascade is affected in human mental retardation syndromes. The mitogen-activated protein kinase (MAPK) family includes two important members that are involved in many different forms of learning and memory. These proteins are termed extracellular-signal–regulated kinases (ERKs) to reflect the fact that they were initially characterized as kinases that became activated after growth factors bound to specific receptors at the outer surface of a cell’s membrane. Upstream regulators and selected potential downstream targets of the ERK MAP kinase cascade are shown, along with known sites of derangement in human mental retardation syndromes. One current model is that MAPK plays multiple roles in memory formation: Modulating the induction of lasting synaptic changes through regulating voltage-dependent potassium channels and triggering long-lasting changes through regulating gene expression via CREB phosphorylation. Other possible sites of action are regulating local protein synthesis, regulating cytoskeletal proteins, and regulating other ion channels such as the AMPA subtype of glutamate receptor. (Adapted with permission from Sweatt JD, Weeber EJ, Lombroso PJ: Cognitive disorders and the ras/ERK/CREB pathway. J Am Acad Child Adol Psychiatry 42:741–744, 2003.). |
Neurofibromatosis is an autosomal dominant disease with clinical features that include neural-derived tumors, and café-au-lait spots throughout the body. Approximately half of the affected individuals are also mentally retarded (14). The gene that causes neurofibromatosis (NF1) was recently characterized and several different mutations were identified in affected patients (15). Variability in the types of mutations (point mutations, insertions, or deletions) reflects the high level of phenotypic heterogeneity seen in affected individuals.
The region of the gene that is mutated determines whether or not the child develops cognitive deficits, in addition to the characteristic benign tumors. In other words, the normal neurofibromin protein has several amino acid domains with distinct cellular functions. One of the domains regulates cellular proliferation, and mutations within this region of the gene lead to the formation of tumors. Another portion of the protein regulates the ERK pathway, and mutations within this region interfere with the ability of neurofibromin to regulate the ERK pathway. As a consequence, the ERK pathway is overactive and does not respond appropriately to incoming neuronal signals. Normal synaptic plasticity does not occur and individuals with this type of mutation are mentally retarded (16).
A mutation in a second protein in the classical ERK pathway leads to Coffin-Lowry mental retardation syndrome (17). One of the downstream targets of the ERK pathway is a kinase called ribosomal S6 kinase (RSK2). RSK2 is a protein kinase that rapidly enters the nucleus upon activation and phosphorylates the important transcription factor, CREB. This transcription factor targets specific genes and induces their expression; in the absence of phosphorylation, CREB remains less active. Mutations in the RSK2 gene disrupt the normal cascade from the neuronal surface to the nucleus, interfere with gene transcription, and proteins that are required for synaptic modifications are not expressed (18).
A third disorder associated with mutations in the ERK pathway is Rubinstein-Taybi syndrome (RTS) (19). RTS individuals have characteristic clinical signs that include facial abnormalities, broad digits, and mental retardation. Recently, a mutation to the CREB binding protein gene (CBP) was discovered in patients with the RTS phenotype. The CBP protein is required for the normal unwinding of DNA that precedes the binding of transcription factors to promoter regions. As a result of the mutation, the transcription factor CREB cannot bind properly to the targeted DNA sequences and gene transcription does not occur. Once again, synaptic plasticity is disrupted and normal learning does not develop.
Fragile X Syndrome
The molecular biology of fragile X syndrome has advanced dramatically over the past decade (20). This disorder is the second most common cause of mental retardation after Down syndrome and affects as many as 1 in 750 to 1,000 males, and 1 in 500 to 750 females (21) (Chapter 5.1.2).
Children with fragile X syndrome are born with mild to severe mental retardation. Additional clinical symptoms include facial, testicular, and connective tissue abnormalities (22,23). Abnormal speech patterns are present in the majority of cases and include echolalia and high-pitched speech, as well as poor articulation, dysfluency, and dyspraxia. There is often gaze aversion among these children, as well as stereotypic behaviors, hyperactivity, and attentional difficulties. Aggressive
and self-injurious behaviors are prominent features in some cases (24). Although there has been some interest in identifying specific linguistic, cognitive, or behavioral deficits among affected individuals, these have not been found (21,25,26,27). This is of some interest given the recent molecular discoveries discussed below indicating a general disruption to synaptic plasticity throughout the brain.
and self-injurious behaviors are prominent features in some cases (24). Although there has been some interest in identifying specific linguistic, cognitive, or behavioral deficits among affected individuals, these have not been found (21,25,26,27). This is of some interest given the recent molecular discoveries discussed below indicating a general disruption to synaptic plasticity throughout the brain.
Fragile X syndrome is transmitted from one generation to the next as an X-linked disorder. It has been known for many years that the phenotype of fragile X syndrome often cosegregates with what appears to be a “fragile” site on the X chromosome. In a certain proportion of cells grown in the absence of the nutrient, folic acid, a break point becomes visible on one of the X chromosomes (Figure 2.3.3.2) (28). The chromosomal region is not in fact separated but rather does not stain normally on karyotype analyses. These findings suggested that the gene or gene(s) involved in the disorder might lie near the disrupted site.
Several unusual aspects of the disorder were noted before the gene was actually cloned. Approximately 20 percent of the males who carry the abnormal gene are not mentally impaired. If the gene lay on the X chromosome, why were these individuals not affected, as they have no normal copy of the gene to compensate for the mutated gene? These men were called “normal transmitting males,” and pass the abnormal gene to their daughters, who may also be unaffected. However, their grandsons are at high risk for the full syndrome. This progression in severity over several generations is known as anticipation.
 FIGURE 2.3.3.2. Fragile X site. The fragile site is shown on an affected chromosome of an individual with fragile X syndrome. (Adapted with permission from Lubs H: A marker X chromosome. Am J Hum Gen 21:231–244, 1969.) |
The molecular basis for anticipation is now understood, and relates to the molecular defect found in the affected FMR-1 gene (29). A novel type of mutation was identified, termed a triplet repeat expansion. Triplet repeats refer to any three bases in the nucleotide sequence that are repeated as a unit several times. In the case of fragile X syndrome, the three repeated nucleotides are cytosine-guanine-guanine (CGG). Normal individuals have between six and 50 repeats of these bases in their FMR-1 gene, with 29 repeats being the most frequent number seen (29). Affected individuals, however, have a dramatic increase in the number of repeated sequences, with 200 to 1,000 repeats typically. Mothers of affected probands often have numbers of CGG repeats that fall in between those seen in normal individuals and those affected with the full fragile X syndrome. Carriers thus typically have between 50 and 200 CGG repeats (29); repeats in this range are called premutations.
Some individuals who carry the premutation of the FMR-1 gene have mild cognitive and behavioral symptoms (30). Heterozygotic fragile X females perform poorly on visuospatial and/or memory subtests (31,32). Moreover, magnetic resonance imaging (MRI) and positron emission tomography (PET) analysis have shown premutation female carriers to have significant decreases in total brain volumes, as well as metabolic increases in the hippocampus and cerebellum (33). Mothers who have the premutation are at a much higher risk of expanding the triplet repeat in the next generation to produce affected children with the full mutation. Although why it happens remains unclear, the expansion change from premutation to the full mutation is the molecular basis for the phenomenon of anticipation.
Two separate phenotypes recently have been described in premutation carriers. Premature ovarian failure is reported to affect 16 percent to 24 percent of female carriers (34). One-third of male premutation carriers over the age of 50 develop fragile X–associated tremor and ataxia syndrome (35,36). The molecular biology of these two distinct disorders remains unclear and does not appear directly related either to the CGG triplet repeat size or to the extent of FMRP deficit.
In addition to the 200 to 1,000 CGG repeats in affected individuals, other abnormalities in the DNA have been noted. CpG islands were discussed in the previous chapter, and are regions often found within promoter regions with high content of the two bases, cytosine and guanosine. Cytosines may be chemically modified by the addition of a methyl group, and large regions of methylated nucleotides regulate whether a gene is expressed or not. The relatively small triplet repeat found in unaffected individuals is present immediately downstream of the promoter region, in a region of the FMR message that does not encode protein (37).
The triplet repeat expansion in fragile X produces a large increase in the number of methylated CpG islands (38). The extent of methylation over the region correlates directly with the loss of functional protein (39). The methylated region also explains the “fragile site” that is observed, as this region of DNA does not stain well on karyotype analyses and appears as a broken chromosomal region. To summarize, the abnormal methylation pattern is adjacent to the promoter region, interferes with normal transcription of the gene, and ultimately leads to the absence of functional protein in most affected individuals.
What is the normal function of the FMR-1 protein and how does its absence lead to clinical symptoms? Soon after the gene was isolated in 1991, researchers noticed that the protein contained three domains with highly conserved amino acid patterns. These domains are homologous to a motif found in proteins that bind to ribonucleic acid (RNA) molecules. These proteins are called RNA-binding proteins and are involved at multiple steps during the processing, trafficking, or
translating of transcripts within cells (40,41,42). A mutation in an RNA-binding protein might seriously impair the ability of cells to produce mature messages or translate those messages into protein.
translating of transcripts within cells (40,41,42). A mutation in an RNA-binding protein might seriously impair the ability of cells to produce mature messages or translate those messages into protein.
This was in fact described in a fragile X patient who did not have the expected triplet repeat expansion, but instead had a point mutation that changed a single highly conserved amino acid normally present within one of the RNA-binding motifs (42,43,44). The domain thus appeared to be critical for the proper function of FMR-1 protein, and mutations in this domain interfere with its ability to bind RNA molecules and to process them correctly. In addition, this case indicated that although the majority of cases are caused by a triplet repeat expansion fragile X syndrome, any mutation that disrupts the functional activity of the FMR-1 protein could reasonably lead to a similar clinical syndrome. It is a good example of allelic heterogeneity, where mutations in different parts of a single gene lead to the identical phenotype.
Knowledge of the intracellular location of the FMR-1 protein has extended our understanding of its function. The fragile X protein binds to ribosomes, the organelles within cells that are made from ribosomal RNA and various RNA-binding proteins and function as factories that translate mRNA transcripts into proteins (45).
Studies of the expression pattern of the FMR-1 gene during development show the areas of the brain that normally have the highest level of expression. These include the basal forebrain and hippocampus (46). Both are involved in short-term memory and sequential processing of information, and are affected in some neurodegenerative disorders, such as Alzheimer’s disease. In addition, clinical neuroimaging studies have detected age-related volumetric changes in fragile X individuals in the cerebellar vermis, fourth ventricle, and hippocampus (47,48,49). Finally, the production of animal models, such as knockout mice, has been helpful in studying the underlying processes that are disrupted. Many of the clinical features seen in fragile X syndrome are present in these knockout mice (51).
A major advance in our understanding of the normal role of FMR-1 has come from two lines of investigation. The first has to do with the question of where proteins are translated within neurons. The older dogma was that messages produced within the nucleus were rapidly transported into the cytoplasm where they attached to ribosomes and deposited the translated proteins into the endoplasmic reticulum for further processing. This is the case for the vast majority of messages. However, within neurons, recent work has suggested that a population of messages is actually transported throughout the dendrites and into the spines themselves. They remain there until an incoming signal arrives and initiates local protein synthesis at the very spines where the proteins are needed.
This new model has been helpful in solving a major difficulty with the earlier dogma. A typical neuron has up to 10,000 spines. Incoming synaptic activity arrives at perhaps several hundred of these spines. The previous model would have had the incoming signal travel to the nucleus, to initiate gene transcription and protein translation in the perinuclear region. The problem that arose was how proteins were then transported to the minority of spines where synaptic modifications were required.
The new model posits that messages themselves are transported throughout the dendrites to the spines, ready for an incoming signal. Evidence for this model has been extensive over the past several years, as various components of the translational machinery have been detected throughout dendrites and within the spines (51). These include the messages themselves as well as ribosomes and various proteins required for local protein synthesis (52).
It should be added that local proteins synthesis does not preclude a signal from being sent from the activated spines back to the nucleus to initiate gene transcription. In fact, these signals must occur for long-term potentiation to be maintained and long-term memories consolidated. There is an initial burst of local protein synthesis that induces long-term potentiation, and a later phase during which mRNA messages must be transcribed to maintain the long-term potentiation. An interesting consequence of this can be demonstrated by injecting transcriptional inhibitors into specific brain regions. If these inhibitors are injected into the amygdala, for example, short-term fear conditioning memories are formed, but this learning is not consolidated into long-term memories, as the maintenance of LTP requires protein synthesis.
Having solved one problem, researchers were left with another. Obviously, the messages that are transported down the neurites must not be translated until the proper synaptic input arrives. Recent work from several laboratories has suggested that inhibition of translation is an important function of FRMP (20). FMRP attaches to newly synthesized mRNAs in the nucleus, accompanies these messages throughout the neuronal network and into spines, all along inhibiting translation of the messages into proteins. When a signal arrives that normally would induce synaptic plasticity, the inhibitory actions of FMRP end and the message is translated into proteins.
Exactly how this happens is not yet understood (53). One interesting possibility is that the kinase ERK is involved. It is now known that one of the functions of activated ERK, besides its ability to transduce signals into the nucleus as described above, is to participate in the regulation of local protein synthesis at the spine itself (54,55). Presumably, these functions of ERK work in parallel: one to initiate rapid local protein synthesis and the second to translocate into the nucleus and activate gene transcription.
The messages that are transported to the spines are a subset of messages found in neurons: messages that encode for proteins necessary for the biochemical and structural modifications occurring at the synapse (56,57). It should be pointed out that FMRP is probably only one of a number of proteins required for this process. Many proteins are needed to support the transport of messages, their inhibition or activation at the spine, as well as providing the scaffolding support for local translation of proteins. Although this is more speculative, it is likely that mutations that disrupt these proteins will be found to also disrupt synaptic plasticity to varying degrees and produce cognitive deficits in affected individuals.
The model presented in the previous paragraphs leads to certain predictions. If FMRP is necessary for synaptic plasticity to develop, then the absence of this key player might result in detectable disruptions in spine morphology. William Greenough and his colleagues have looked at this question and have found evidence supporting the hypothesis. They used anatomical techniques to examine spines from patients with fragile X syndrome as well as a mouse model of the syndrome (58,59). Compared to the controls, they found larger numbers of long, spindly, and immature-looking spines in the fragile X cases, and fewer numbers of short, mushroom-shaped, and mature-looking spines. Taken together, these studies suggest that FMRP is involved in development of spines through regulation of the shape, number, and maturity of spines, and that this occurs through FMRP’s regulation of local protein synthesis.
Expansion of the FMR-1 gene through triplet repeats was originally thought to be a genetic anomaly unique to fragile X syndrome. More than a dozen other triplet repeat disorders have now been identified (Figure 2.3.3.3), including Huntington’s chorea, Friedreich’s ataxia, and myotonic dystrophy (60,61,62). The phenomenon of anticipation had been apparent in most of these disorders but early investigators dismissed it as the result of ascertainment bias. The discovery of triplet repeat expansions, however, provided the molecular
explanation for this phenomenon. The degree of anticipation can be quite remarkable (Figure 2.3.3.4). In myotonic dystrophy, for example, expansions that are just above the threshold for disease may only result in individuals developing cataracts late in life. Several generations later, descendants have the full expansion with long repeats and fatal congenital illness (63). Interestingly, anticipation has been described in families with schizophrenia and bipolar disorder, and some investigators believe triplet repeat expansions may account for a subset of these affected patients (64,65,66,67,68).
explanation for this phenomenon. The degree of anticipation can be quite remarkable (Figure 2.3.3.4). In myotonic dystrophy, for example, expansions that are just above the threshold for disease may only result in individuals developing cataracts late in life. Several generations later, descendants have the full expansion with long repeats and fatal congenital illness (63). Interestingly, anticipation has been described in families with schizophrenia and bipolar disorder, and some investigators believe triplet repeat expansions may account for a subset of these affected patients (64,65,66,67,68).
Related posts:
 Child and Family Policy: A Role for Child Psychiatry and Allied Disciplines
Oppositional Defiant and Conduct Disorders
Child and Family Policy: A Role for Child Psychiatry and Allied Disciplines
Oppositional Defiant and Conduct Disorders
 Interpersonal Psychotherapy
Interpersonal Psychotherapy
 Understanding Research Methods and Statistics: A Primer for Clinicians
Suicidal Behavior in Children and Adolescents: Causes and Management
Integrating Behavioral Services into Pediatric Care Settings: Principles and Models
Understanding Research Methods and Statistics: A Primer for Clinicians
Suicidal Behavior in Children and Adolescents: Causes and Management
Integrating Behavioral Services into Pediatric Care Settings: Principles and Models
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


