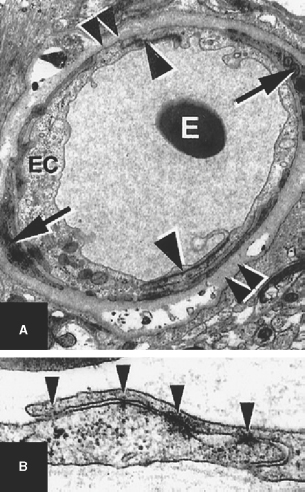
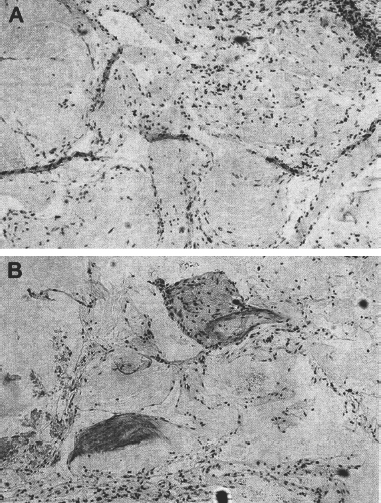
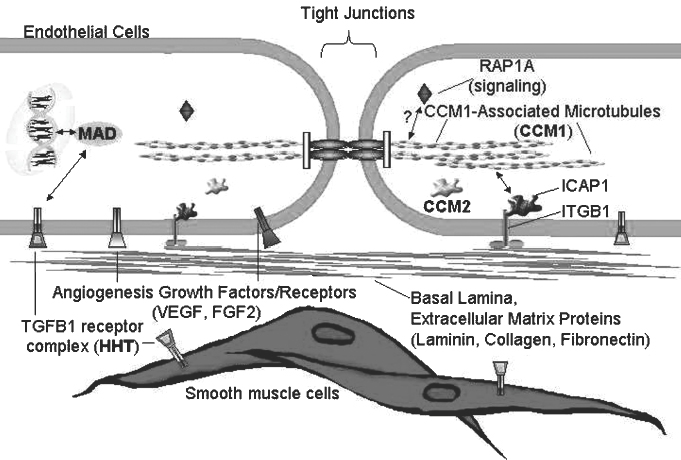
2 Pascal M. Jabbour, Judith Gault, Robert Shenkar, and Issam A. Awad Cerebral cavernous malformations (CCMs) are proliferative hemorrhagic lesions consisting of clusters of sinusoidal caverns lined by endothelium and filled with thrombus at various stages of organization.1 The lesions are compact and surrounded by gliosis and blood degradation products. Their prevalence in the general population is estimated to range from 0.1 to 0.9%.1–8 Depending on their size and location, the lesions can be clinically silent or manifest symptoms ranging from headaches and focal neurologic deficits to seizures and severe intracranial hemorrhage.2–9 Ultrastructural analysis suggests abnormalities of the interendothelial tight junctions and subendothelial layer of the blood-brain barrier.10–12 Typically, lesions are confined to the central nervous system, occurring in a volume distribution,5 but they rarely involve the epidural space and other locations including the skin, liver, and orbit.13–15 The CCMs occur in a sporadic or autosomal dominant inherited form. Familial cases are characterized by the multiplicity of lesions, whereas sporadic cases harbor usually a single overt CCM.16–18 The frequency of the familial form has been estimated to be as high as 50% in Hispanic-American patients17,19 and 20 to 30% in other patients.19 Three CCM loci have been identified: CCM1 on 7q, CCM2 on 7p, and CCM3 on 3q.16, 20–22 The disease gene responsible for CCM1 encodes the Krev interaction trapped 1 (KRIT1) protein,16,23,24 and recently the CCM2 gene, MG4607, has been identified.25 Current lesion nomenclature of cerebral vascular malformations is based on classic histologic descriptions by Russell and Rubinstein.26–29 The arteriovenous malformations (AVMs) exhibit mature vessel wall elements with direct communications between arteries and veins and a high-flow profile predisposing to vascular recruitment, arterialization of venous structures, and gliosis of intervening and adjacent brain tissue. They have three major components: arterial feeders, nidus, and venous outflow. The AVMs are prone to apoplectic hemorrhage by rupture of nidal vessels or associated aneurysms or by venous outflow obstruction.26,29,30 In contrast, the CCMs appear to grow by a process of vascular cavern proliferation, in the setting of repetitive lesional hemorrhages. The CCMs exhibit brittle vascular morphology devoid of mature vessel wall elements.31,32 They do not exhibit the high-flow features of AVMs and are less commonly associated with apoplectic hemorrhage.5,7,33,34 A third and the most common form of cerebral vascular malformation is the venous malformation (VM), also known as venous angioma or venous developmental anomaly, a lesion that rarely manifests clinical sequelae except when associated with a CCM lesion.35,36 It is composed of abnormally enlarged vessels of venous structure separated by normal neural parenchyma.8,37 These vessels are arranged in a radial pattern extending from a dilated central venous trunk, which itself drains into either a deep or superficial venous sinus.37,38 Large VMs often reflect regional venous dysmorphism and serve as sole or major channels of venous drainage of adjacent brain. Mixed vascular malformations, exhibiting features of more than one lesion type, have been described.39 Nonfamilial CCMs are often associated with a VM in the same region of the brain, whereas this association is rare in familial cases.35 The CCM lesions are often associated with adjacent nonhemorrhagic capillary telangiectasia (mixed CCM-capillary malformations), and components of CCM lesions may exhibit maturation of vessel wall, akin to true AVMs. Early biologic studies and genetic investigations have largely considered and compared pure AVMs and CCMs and avoided the mixed lesions. Future studies will need to explore and compare the mixed vascular malformations in comparison with the pure forms, as their pathobiology will likely shed critical light on the nature of individual lesion types and their pathogenesis and evolution. Other vascular malformations including the dural AVMs, with arteriovenous shunting within the dural leaflet, are also important clinically, and there has been new information on their pathobiology. However, consideration of these lesions is beyond the scope of this review. Figure 2-1 Brain magnetic resonance imaging (MRI): Gradient-echo-weighted image showing multiple cerebral cavernous malformations (CCMs). Only the larger symptomatic infratentorial lesion (large arrowhead) was visible on conventional MR sequences (T1, T2, and FLAIR [fast fluid-attenuated inversion-recovery]), whereas the multiple supratentorial “baby lesions” (small arrows) were only seen on gradient-echo imaging. Magnetic resonance imaging (MRI) is considered the gold standard for the diagnosis of CCMs,5,40–48 with typical features of mixed signal intensity within the lesion and a rim of surrounding hypointensity reflecting hemosiderin.49,50 Gradient-echo MRI can detect small areas of chronic hemorrhage not seen on conventional spin-echo techniques, including multiple minute lesions in familial cases thought to represent precursors of larger and more symptomatic lesions (Fig. 2-1). MRI with contrast is also important to detect any venous malformation associated with the CCM. The clinical manifestations of CCMs include incidentally discovered lesions, those presenting with headache or nonspecific symptoms, seizures, hemorrhage, and focal neurologic deficits. Severity of disease is mostly defined in relation to the frequency of clinically overt hemorrhagic episodes. Other indices of disease severity include clinical penetrance in familial cases (frequency of affected kindreds), age at clinical presentation, prevalence of clinical disability, and the need for surgical interventions.9,14 Most clinical series include 14 to 19% of cases with asymptomatic lesions incidentally discovered on MRI.5,51,52 The prevalence of overt hemorrhage ranges between 6% and 30% of cases.5,51,53–56 The risk of overt hemorrhage is estimated to range between 0.25% and 0.7% per lesion per year,57–61 with infratentorial lesions significantly more likely to cause clinically significant and disabling bleeds.62 Solitary lesions are most common in the sporadic form of the disease, occurring in 60 to 80% of cases. Multiple lesions are the hallmark of familial cases, occurring in increasing frequency with age and more likely detected by gradient-echo MRI.6, 63–70 De novo CCMs appear commonly in familial cases,71 with a rate of new CCM formation on MRI of 0.4 to 1 lesions per patient-year.63 De novo formation of CCMs has also been documented in the setting of preexisting VM and after cranial radiation therapy to treat an unrelated lesion and in the absence of any other apparent causative factors.72–75 Mixed lesions (mixed VM-CCM, mixed AVM-CCM, and mixed VM-AVM) have been described, which include features of more than one lesion type. It is not clear if these develop as mixed or transitional lesions or instead evolve mixed features from preexisting malformations. The mixed VM-CCM has been well described19,32,76 in 20 to 30% of CCMs. Familial CCMs are rarely associated with VM. The AVM-CCM association has largely been reported in pathologic specimens of lesions presenting as angiographically occult vascular malformation, indistinguishable clinically and radiographically from pure CCMs. One possible explanation is that microhemorrhage from a cryptic AVM results in reactive angiogenesis and ultimately the formation of a CCM.33 Another possibility is that some CCMs include portions with attempted vessel maturation, with features of mature vessel wall elements mimicking AVM.77 The mixed VM-AVM is the least common.78,79 One proposed explanation is that the VM may be a potential precursor of AVMs with fistulization of arteriovenous bed draining into a VM.78 Blood vessels within central nervous system parenchyma are composed of a tunica intima, media, and a specialized outer layer intimate with surrounding astrocytic milieu.80 The tunica intima maintains a competent blood-brain barrier and consists of a layer of endothelial cells connected by specialized tight junctions, resting on a basal lamina including pericytes and specialized matrix proteins fibronectin, laminin, and collagen IV, that provide structural support and anchor endothelial cells32,81 (Fig. 2-2). Astrocytic foot processes provide additional structural integrity and unique signaling opportunities. Generally, immature vessels early in angiogenesis consist of nonadherent proliferating endothelium in a fibronectin-rich matrix, whereas more mature vessels have an adherent nonproliferating endothelium and express laminin more uniformly with a paucity of fibronectin in the matrix.81 Collagen IV is abundant within the basal lamina and important in maintaining structural integrity of the vessel wall.80 The tunica media consists chiefly of concentric layers of helically arranged smooth muscle cells; interposed among smooth muscle cells are variable amounts of elastic and reticular fibers, in addition to proteoglycans.80 The tunica media layer is rich in extracellular matrix protein, collagen III, expressed at all stages of angiogenesis, and various vascular smooth muscle cell proteins including, α-smooth muscle actin, myosin heavy chain, and smoothelin (SMTN) are expressed in this layer.32,82 In arteries, the media is separated from the intima by an internal elastic lamina, a layer largely composed of elastin protein. Larger arteries possess a thinner external elastic lamina that separates the media from the outer tunica adventitia. In capillaries and postcapillary venules, the media is represented by pericytes. Figure 2-2 Transmission electron micrographs of normal cerebral capillary. (A) Pericytes (arrows) within the basal lamina (double arrowheads); EC, endothelial cell; arrowheads, tight junctions; E, erythrocyte (original magnification, × 8100). (B) Normal endothelial cells. Arrowheads, tight junctions (original magnification, × 42,000). (From Wong JH, Awad IA, Kim JH. Ultrastructural pathological features of cerebrovascular malformations: a preliminary report. Neurosurgery 2000;46(6):1454–1459. Reprinted with permission.) Vascular smooth muscle cells are integral to vascular repair and provide structure to the tunica media and vessel wall. Of the two vascular smooth muscle phenotypes, synthetic and contractile, the latter is more common in the adult population. α-Smooth muscle actin is a contractile protein found earliest in smooth muscle cell differentiation; it is the most abundant contractile protein in vascular smooth muscle cells. Actin appears to be expressed in all layers of arteries and in the subendothelium (intima) of veins. Myosin heavy chains (MYHCs) are ubiquitous actin-based motor proteins that convert the chemical energy (ATP) into mechanical force involved in cytokinesis, vesicular transport, and cellular locomotion. In eukaryotic cells, myosin heavy chains (MYH1 and MYH2) are present at a later stage of smooth muscle differentiation than actin and are expressed in all layers of arteries but not in veins. SMTN is a cytoskeletal protein that is different from other smooth muscle-specific proteins, which are expressed in both synthetic and contractile smooth muscle cells; SMTN is specific for contractile cells and serves as a marker for these cells. SMTN is expressed in the media of some arteries but not in veins.32,80,81,83 Figure 2-3 Transmission electron micrographs of CM. (A) Fibrous septa separating caverns at top and bottom; E, erythrocyte; arrowheads, endothelial cells; arrows, subendothelial cells; A, amorphous connective tissue; CM, cavern wall; C, collagen bundles (original magnification, × 4700). (B) Peripheral capillary showing electron-dense hemosiderin (H) ring with tight junction (arrowhead) (original magnification, × 7600). (C) CM, cavern wall; EC, endothelial cell; P, pericyte; S, subendothelial cells; arrowheads, basal lamina (original magnification, × 5100). (D) Lack of normal interendothelial cell junctions (arrows); double arrowheads, basal lamina. Top, original magnification, × 100,000; middle, original magnification, × 84,000; bottom, original magnification, × 69,000. (From Wong JH, Awad IA, Kim JH. Ultrastructural pathological features of cerebrovascular malformations: a preliminary report. Neurosurgery 2000;46(6):1454–1459. Reprinted with permission.) CCMs are characterized by caverns that are filled with blood or thrombus, lined by a single layer of endothelial cells, and separated by an amorphous connective tissue matrix (collagen).11 The lining of caverns in CCM lesions lacks interendothelial cell tight junctions and subendothelial support11,12 (Fig. 2-3). Endothelial tight junctions are a component of the blood-brain barrier, and their disruption is consistent with an underlying abnormality in cytoskeletal structure. The CCM subendothelium, at the level of the basal lamina (extracellular matrix), expresses fibronectin to a greater extent than normal brain vessels and AVMs. In contrast, laminin is underexpressed in the subendothelial layer of CCMs compared with AVMs and normal vessels. The CCMs express collagen IV within the subendothelial layer and collagen III only focally in the perivascular tissue. Smooth muscle maturation in CCMs has recently been studied82 (Fig. 2-4). Most caverns in CCMs express actin in the subendothelial layer. Only 20% of large caverns express MYHC in the subendothelial layer, and most small caverns do not express MYHC. Actin and myosin expression in CCMs is largely limited to the subendothelial layer, with scant expression within the intercavernous matrix. The CCM lesions do not exhibit recognized muscularis layers as in normal vessels and AVMs. SMTN expression, characteristic of contractile smooth muscle cells, is absent in CCMs.82 The CCMs have characteristic hemosiderin deposits near the basal lamina and lack astrocytic foot processes; the processes stop at the border of the lesion.11,12 Figure 2-4 Immunohistochemistry of [α]-SMA, myosin heavy chain (MYHC), and smoothelin (SMTN) in cerebral cavernous malformations (CCMs). (A) Negative control (original magnification, × 100). (B) Immunostaining for [α]-SMA showing confined delineation of walls of the caverns. The staining involved the subendothelial cell layer and rare scattered cells in intercavernous connective tissue (original magnification, × 100). (C, D) Immunostaining for MYHC showing immunoexpression in the thin subendothelial layer of some but not all caverns. Note many caverns devoid of MYHC expression in their walls. (D) MYHC expression was localized in the subendothelial layer (original magnification, × 400). (E) Immunostaining for SMTN showing no expression in any caverns (original magnification, × 100). (From Uranishi R, Baeu NI, Kim JH, et al. Vascular smooth muscle cell differentiation in human cerebralvascular malformations. Neurosurgery 2001;49(3):671–680. Reprinted with permission.) Figure 2-5 CM, VEGF expression. (A) control; (B) anti-VEGF. Note the staining of the intercavernous matrix and the endothelium. (From Rothbart D, Awad IA, Lee J, et al. Expression of angiogenic factors and structural proteins in central nervous system vascular malformations. Neurosurgery 1996;38(5):915–925. Reprinted with permission.) Normal adult cerebral vasculature generally does not express angiogenesis factors such as vascular endothelial growth factor (VEGF) and fibroblast growth factor 2 (FGF2). VEGF, FGF2, and TGFB1 expression in cerebral vascular malformations is possibly induced by proliferation of new vessels, hemodynamic stress, ischemia, and/or hemorrhage.32,84 VEGF is predominately expressed in the subendothelial layer and media of AVM vessels and in the intercavernous matrix and subendothelial layer in CCM lesions32 (Fig. 2-5). FGF2 is expressed in the media of AVM vessels and in both the subendothelial layer and the intercavernous matrix of CCMs.32 The proportion of fmsrelated tyrosine kinase 1 (FLT1) and kinase insert domain receptor (KDR) immunopositive vessels is significantly greater in AVMs and in CCMs compared with control brain (P < 0.05).85 Increased tyrosine kinase receptors (TEK) are detected in AVMs and CCMs compared with control brain (not statistically significant), and tyrosine kinase with immunoglobulin (TIE) was detected in rare vessels of all lesion types and brain.85 Compared with CCMs and AVMs, VMs rarely hemorrhage and are variants of normal venous drainage. Kilic et al. described two VMs with negligible expression of angiogenic factors VEGF, FGF2, and TGFB suggesting they are angiogenically dormant.84 Families with CCM exhibit Mendelian inheritance at three chromosomal gene loci (CCM1, CCM2, CCM3). The CCM1 gene was positionally cloned by linkage, haplotype, and mutation analyses mainly in CCM families with a Hispanic-American ancestral disease haplotype24,86 in the 7q11.2-q21 region and a common mutation in CCM1 that encodes the KRIT1 (Krev interaction trapped 1) protein.21, 24,87,88 The CCM1 gene consists of 20 exons spanning 45,799 base pairs (bp) and maps to the 7q11.2-q21 region. The start of translation appears to be in exon 5, and more than 88 different germline mutations distributed throughout the CCM1 gene have been described in association with CCM in many different races.13,23,24,89–91 Mutations in CCM1 described to date all presumably result in CCM1 protein truncation. Germ-line CCM1 mutations have been identified in apparently sporadic CCM cases with multiple lesions that were due to unrecognized familiality or spontaneous germ-line mutations.86,92 A novel gene, MGC4607, exhibiting eight different mutations was recently identified in nine families with linkage to the CCM2 locus.16,25 Two different somatic mutations in CCM1 were identified in DNA isolated from a lesion surgically excised from a patient without a family history of the disease and harboring a single CCM, supporting a two-hit hypothesis of lesion genesis.93,94 Another study performed by Reich et al.95 failed to identify CCM1 somatic mutation in 72 paraffin-embedded CCM lesions suggesting the role of somatic mutation requires further investigation. In the two-hit model, a vascular cell with two mutations (either germ line or somatic), resulting in complete loss of functional CCM1 protein, clonally expands to form a CCM lesion. Presumably, the multiple CCM lesions found in familial cases result from the same germ-line mutation found in every cell plus a different somatic mutation. Multiple lesions would not be the result of metastasis. Familial CCM exhibits an autosomal dominant mode of inheritance but is likely recessive at the cellular level, and lesion genesis may require two hits to the same gene. Since the identification of the CCM1 gene, investigations have been focused on functional characterization of the CCM1 protein. Three functional domains have been predicted based on sequence homology with known proteins and protein-protein analysis using yeast two-hybrid screening. The two-hybrid system is set up to detect transcription of a reporter gene either by colormetric tests or selection for growth. Reporter gene transcription depends upon association of a DNA-binding domain, fused to a gene of interest, and a transcriptional activation domain fused to many different genes that may interact with the gene of interest. Protein-protein interactions are identified when a yeast colony grows or turns blue indicating the fusion protein containing the gene of interest is interacting with one of the fusion proteins containing the activation domain and allowing transcription of the reporter gene. The NPXY motif, in the amino terminal of CCM1, apparently binds integrin cytoplasmic domain-associated protein (ICAP1), suggesting CCM1 is part of the integrin signaling pathway (specifically with β1-integrin complexes) and cell adhesion to other cells as well as to the extracellular matrix.87,96,97 At least 8 of 22 integrin heterodimers are expressed on angiogenic and quiescent vascular endothelial cells including five with β1-integrin components.98 Integrins are activated by VEGF and FGF2 signaling in angiogenesis.98,99 Ankyrin repeats, in the middle of the CCM1 protein, are thought to be involved in protein-protein interaction. The FERM domain found in exons 14 to 18 of CCM1 has been found in proteins that link cytoplasmic proteins to transmembrane proteins. The carboxy-terminal of CCM1 interacts with Ras-related protein 1A (RAP1A) (alias Krev-1/Rap1a), a member if the Ras-family of GTPases, using a yeast two-hybrid screen suggesting a tie to the tumor suppression pathway possibly at the point where Ras becomes part of the integrin pathway.87,97,100 The CCM1 gene has an alternately spliced form missing the region required for Rap1a.2 The CCM1 gene is transcribed as a 3.5-kb message in brain and additional tissues not known to be affected in CCM patients (heart and muscle).87,94,101 Recently, published data from Guzeloglu-Kayisli et al.10 demonstrated that KRIT1 is expressed in a broad variety of human organs, where it localizes to the vascular endothelium of capillaries and arterioles of each organ. KRIT1 antibodies stained intensely where endothelial cells tend to adhere to one another like the blood-brain barrier, suggesting that additional cell type contributes to the pathophysiology of CCMs. Using CCM1 antibodies, CCM1 colocalizes with β-tubulin in endothelial cells in culture and is thought to interact with the cytoskeleton to determine cell shape through cell-cell and cell-matrix interactions.102 Whitehead et al. demonstrated using murine gene targeting that CCM1 is required for vascular development, and KRIT1-associated vascular defects are not secondary to disrupted neural patterning but secondary to disrupted genetic pathway important in establishing arterial identity.103 The CCM2 gene encodes a protein with a phosphotyrosine-binding domain, similar to ICAP, and may lead to the formation of CCM by a similar mechanism involving altered regulation of integrin-mediated intercellular tight junctions.16,25 The majority of patients (50 to 80%) with CCM are apparently sporadic without a known family history of CCM.7,11 Single CCM lesions may be found in roughly 75% of sporadic cases and 8 to 19% of familial cases.11,19,104,105 Examination of 202 cases with CCM1 mutations found 13% of cases with a single CCM lesion and 2.5% with no detectable lesion using T2-weighted MRI.19 The presence of multifocal lesions is indicative of familial forms of CCM. Conversely, solitary lesions and lesions associated with VM are less likely associated with familial disease. Familial CCMs have been associated with gene defects likely impacting on endothelial cell cytoskeleton, and this presumably causes defects in interendothelial cell adhesions (Fig. 2-6) and defective maturation of proliferating capillaries in response to ongoing hemorrhage. It is likely that sporadic lesions reflect a disruption involving the same respective genes within the lesions themselves (somatic mutations) or disruption of the same molecular pathways resulting in similar alteration of signaling and ultimately the same lesion phenotype as in familial cases. Hence, the genetic story of familial cases can hold the key to revealing molecular pathways for the genesis of familial and sporadic lesions.
Molecular Biology of Cerebral Cavernous Malformations
 Characterization of Cerebral Cavernous Malformations
Characterization of Cerebral Cavernous Malformations
Types of Vascular Malformations in the Central Nervous System
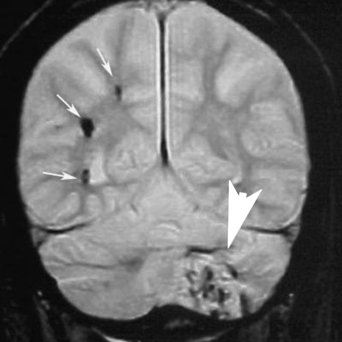
Clinical Variants of Cerebral Cavernous Malformations
Cerebral Cavernous Malformations and Mixed Vascular Malformations
 Histopathology and Ultrastructure of Normal Vasculature
Histopathology and Ultrastructure of Normal Vasculature
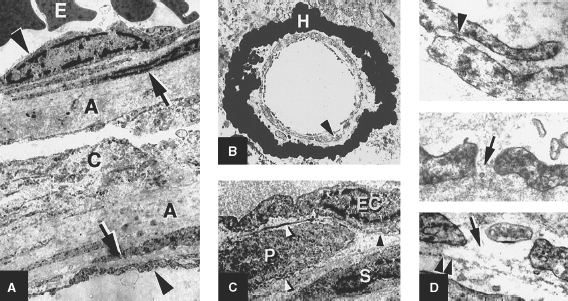
 Angioarchitecture of Cerebral Cavernous Malformations
Angioarchitecture of Cerebral Cavernous Malformations
Ultrastructure of Cerebral Cavernous Malformations
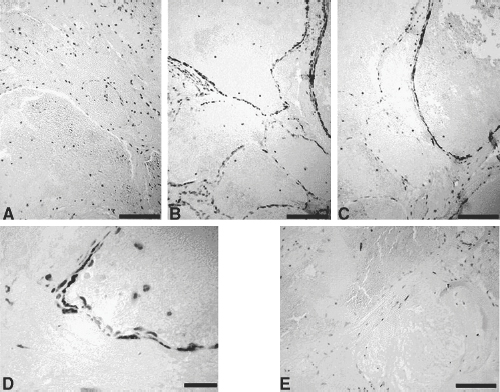
Angiogenesis Activity in Cerebral Cavernous Malformations
 Genetics of Cerebral Cavernous Malformations
Genetics of Cerebral Cavernous Malformations
Genetic Substrate of Familial Cerebral Cavernous Malformations
The Two-Hit Hypothesis of Lesion Genesis
Cerebral Cavernous Malformation Gene Functions
Sporadic versus Familial Cases of Cerebral Cavernous Malformations
Related posts:
 Neuroradiology and Classification of Cavernous Malformations
Neuroradiology and Classification of Cavernous Malformations
 Deep-Seated Cerebral Cavernous Malformations
Deep-Seated Cerebral Cavernous Malformations



